0022-538X/83/120591-13$02.00/0
Copyright © 1983,American Society for Microbiology
High-Resolution Characterization of Herpes Simplex Virus
Type
1
Transcripts Encoding Alkaline Exonuclease
and
a
50,000-Dalton Protein Tentatively Identified
as a
Capsid
Protein
R. H.COSTA,1 K. G.DRAPER,1 L. BANKS,2 K. L. POWELL,2 G. COHEN,3R. EISENBERG,4 AND E. K.WAGNER'*
Department of Molecular Biology and Biochemistry, University of California, Irvine, California 92717'; Department ofMicrobiology (Virology), University of Leeds, Leeds LS2 9JT, England2; and Department of
Microbiology and Center for Oral Health Research, DentalMedicine,3andDepartmentof Pathobiology, Schoolof Veterinary Medicine,4 University of Pennsylvania, Philadelphia, Pennsylvania 19104
Received 27 June 1983/Accepted 9 September1983
Fourpartially overlapping mRNAs (1.9, 2.3, 3.9, and 4.5 kilobases [kb])were
located between 0.16 and 0.19 map units on the herpes simplex virus type 1
genome. Their direction oftranscription was foundto be fromrightto left. The
2.3-kbmRNAwasfoundtobeearly(1),whereas the otherswerelate
(P3y).
Partialsequence analysisof the DNA encoding thesegenes indicated that the promoter
for the2.3-kb mRNA shares structural features with otherearly(1)promoters. In
vitrotranslation of hybrid-selectedmRNA indicated thatamongtheproteinsthese
mRNAs encodeare an82,000-dalton (d) polypeptidereactive withamonoclonal
antibodyagainst herpes simplexvirustype 2 alkaline exonucleaseanda50,000-d polypeptide weakly reactive with apolyclonal antibodymade against thecapsid protein VP19C. Furtherexperiments suggested that the 2.3-kb mRNA encodes the82,000-d polypeptide,whereasone(or both)of thelargermRNAsencodes the
50,000-dprotein. Anovelfindingwasthat the 1.9-kb mRNAappearstosharepart
of the translationalreadingframe for alkalineexonuclease,butanypolypeptideit
encodes does not reactwith the monoclonalantibody to thisenzyme.
We have established in a number of recent publications (reviewedinreference 35)that her-pes simplex virustype 1 (HSV-1) mRNAs map relatively simply on the viral genome. Further-more, the low frequency and relatively short spanofthecharacterized HSV-1 intronssuggest that the correlation ofspecific transcripts with genetic markers and known viralproteins willbe generallystraightforward. Thus, HSV transcrip-tion maps add a valuable further dimension to high-resolution genetic marker localization.
As described in the most recent available genetic mapping report (S. K. Weller, W. R. Sacks, D. M. Coen, and P. A. Schaffer, Virolo-gy,in press), five complementation groups map in the region between 0.1 and 0.2 mapunits on
theParrangementof the HSV-1 genome. Details ofthe datamappingthese groupsarepresented in thatpublication. Twocharacterized viral pro-teins have been located in thisregion. The first is alkaline exonuclease, an important enzyme of HSV-induced DNA replication (10, 16, 25, 26, 33). Preston andCordingley (29) mapped this by measuringenzyme levels of in vitro translation productsofhybrid-selected mRNAandby
per-forming hybrid-arrestedtranslation. The second viral protein characterized isa50,000-dalton (d) capsid protein (VP19C) located byLemasterand Roizman (20),using intertypic recombinants.
Recent work (L. Banks, D. J. M. Purifoy, R. A. Killington, and K. L. Powell, J. Gen. Virol., in press) has led to the isolation of several monoclonal antibodies against HSV-2 alkalineexonuclease, some of whichare
cross-reactivewith theHSV-1-inducedenzyme. Previ-ously reported work (5) described the prepara-tion ofpolyclonalrabbit antiserareactivewitha
50,000-d capsid protein (NC-2 in that publica-tion), which can be inferredto be VP19C. This protein appears to be located on the capsid vertices (34).
Inthe presentstudy,weused theseantiserato
identifythe mRNAencodingthealkaline
exonu-cleaseas a 1 (early) one and, tentatively, a
Py
(late) mRNA as encoding the capsid protein. High-resolution mapping of these mRNAs indi-cated thatall are partially colinear. Correlation oftranscript locationwith nucleotide sequence data demonstrated that the 5' end of the , mRNA lies downstream of a 120-base region
591
on November 10, 2019 by guest
http://jvi.asm.org/
Downloaded from
on November 10, 2019 by guest
http://jvi.asm.org/
Downloaded from
on November 10, 2019 by guest
http://jvi.asm.org/
sharingcertain features with promoters for other ,B HSV-1 mRNAs.Another mRNAwasfoundto
be colinear with the 3' portion of the alkaline exonuclease mRNA. It appeared to have its own independent 5' end and was temporally a
3-y
mRNA. Preliminary data suggested that this mRNA can be translated in vitro toyield
a polypeptide smaller than alkaline exonuclease. Such apolypeptide was not reactive with theQl antibody, but partial sequence analysis suggest-ed that it is encoded by the same translation frame as the alkaline exonuclease.MATERIALSANDMETHODS
Cellsandvirus. ForRNApreparation,
plaque-puri-fied isolates of the KOS strain of HSV-1 were usedto
infect HeLa cells. Monolayer cultures of HeLa cells
weregrown at 37°C in Eagle minimal essential medium
containing 10% calf serum, penicillin, and
streptomy-cin. HSV-2alkaline exonuclease was prepared from
human epidermoid carcinoma no. 2 (HEp-2) cells
grownin thesamemedium. HSV-2wasthe 186strain.
Enzymes. All restriction enzymes were obtained
from Bethesda Research Laboratories. Digestions
were carried out in buffers recommended by the
supplier. Phage T4 polynucleotide kinase (Bethesda
ResearchLaboratories)wasusedfor 5' endlabelingas
described byMaxamand Gilbert(23). Escherichiacoli
DNA polymerase I (Klenow fragment,
Boehringer-Mannheim) was used to generate 3'-end-labeled
re-strictedDNAbyprocedures described by Maniatiset
al(21).
Isolation, labeling,andsizefractionation of
polyribo-somal RNA.Monolayer cultures ofHeLacells(2x107
cells perflask)wereinfected for30minat a
multiplic-ityof10PFUof virus per cell inphosphate-buffered
saline containing 0.1% glucose and 1.0% fetal calf
serum. Polyribosomes were isolated from the
cyto-plasm ofHSV-1-infected cells by themagnesium
pre-cipitation method of Palmiter (27). Polyadenylic
acid-containing [poly(A)] mRNA was isolated from total
rRNAbyoligodeoxythymidylicacid-cellulose
(Collab-orative Research, Inc.) chromatography. This is
re-ferred to as HSV poly(A) mRNA. Details of this
procedurewere presented elsewhere (13). RNA was
isolated at 6 h postinfection except when early (,)
RNA wasrequired. Then cellswerepreincubatedfor1
h andtreated for 5 h with 1.5 x
1i-'
M adenosinearabinoside and 3.7 x 106 M pentostatin to inhibit viral DNA replication, as described previously (15). RNA wassizefractionatedbyelectrophoresison1.4%
agarose gels containing 10 mM methylmercury
hy-droxide(3)as previously described (1, 2).
Recombinant DNA. All recombinant DNA clones
described in this paper were derived from either
HindIIIfragment IO(map units0.082-0.182),HindlIl
fragment J (0.182-0.261). XhoI-HindIII fragment
C'-10 (0.171-0.182), BamHI-HindIII fragment A-IO
(0.152-0.182), or HindIII-SalI fragment J-D
(0.182-0.195), cloned in pBR322. Procedures for cloning
HSV-1 DNA fragments in the pBR322 vector were
described previously (1, 6). Cloned DNA fragments
were named asdescribed previously andlocated by
their map coordinates on the P arrangement ofthe
HSV-1 genome(6).
In situ Northern RNAblots. As described
previous-ly, samples (5 ,ug) of HSV poly(A) mRNA were
fractionated on methylmercury gels and dried onto
Whatman3mm paper with vacuum (15). The agarose
film was floated off the paper in water and hybridized
with appropriate nick-translated, 32P-labeled DNA
probesin50% formamide containing 0.4MNa+, 0.1 M
HEPES
(N-2-hydroxyethylpiperazine-N'-2-ethanesul-fonic acid; pH 8.0), 0.005 M EDTA, and Denhardt
solution (7)at50°C for 36 h. Blots were rinsed at 50°C:
thefirst two rinses were in50%
formamide-2x
SSC(1x SSC=0.15 MNaCl plus 0.015 M sodium
citrate)-0.1% sodium dodecyl sulfate (SDS). The last rinse was
in 0.1x SSC-0.1% SDS. Autoradiography was on
Kodak XRPfilm withorwithout intensifying screens
asneeded.
Invitro32P-labeledDNA wasmade by
nick-translat-ing appropriate DNA clones with DNA polymerase I,
DNase I (Boehringer-Mannheim), and 50 ,uCi of
[a-32P]dCTP(3,000Ci/mmol, Amersham).
Isolation of restrictionfragment-specific mRNA.
Re-strictionfragment-specific mRNA was isolated from
HSV poly(A)mRNAby preparative hybridization to
theappropriate DNA covalently coupled to cellulose.
Details ofcouplingofDNA tocellulose and
prepara-tivehybridizationwere asdescribed previously (2, 6).
Nuclease mappingofHSV-1mRNA.S1nuclease and
exonuclease VII analysis of RNA was carried out
essentiallyasdescribedby Berk and Sharp (4) andas
describedpreviously (1, 6, 8, 11-13, 15). Appropriate
HSV-1 DNA clones (10 ,ug) were cleaved at the
desired site with the appropriate restriction enzyme.
TheDNAthenwas5' labeled with[-y-32P]ATP(3,000
Ci/mmol, ICN), usingpolynucleotidekinase (Bethesda
ResearchLaboratories), toaspecific activity of 4,000
to 10,000 cpm/,ug of DNA. Alternatively, the DNA
was 3' labeled tothesame specific activity by using
DNA polymerase I-Klenow fragment
(Boehringer-Mannheim).
The DNAfragments thenweredenatured and strand
separated on 5% acrylamide gels as described by
Maxam and Gilbert (23). The strand-separated DNA
(from 10 ,ug of cloned DNA)washybridized with 10
jig
of infected-cell mRNA in 0.1 M Na+-0.1 M HEPES
(pH8.0)-0.01MEDTAat65°C for6 to16hina30-,u
volume.Hybrids were subjected toS1nuclease
(Boeh-ringer-Mannheim)or exonuclease VII (Bethesda
Re-search Laboratories) digestion as described (11, 12).
Materialwasfractionatedon adenaturing 5%
acryla-midegel with 5'-end-labeled, HaeII-orHinfI-digested
pBR322DNAfragmentsas sizestandards. When the
nuclease-protected materialwas tobe fractionatedon
sequenceladders, mung beannuclease(PL
Biochemi-cals)wasused insteadofS1 nuclease. Allprocedures
were basedon thoseofMaxamandGilbert(23).
Nucleotidesequencing. As describedpreviously (8,
13), nucleotide sequenceanalysis was carriedoutby
theprocedures ofMaxamand Gilbert(23).
Invitrotranslation.Translation of size-fractionated
viral mRNA was carried out in vitro by using a
micrococcal nuclease-treated rabbit reticulocyte
sys-tem(NewEnglandNuclearCorp.)with
[35S]methion-ine (>800 Ci/mmol) as the radioactive amino acid.
Detailsof theprocedureandfractionation of
polypep-tides in SDS-acrylamide gels by the method of
Laemmli(19) weredescribed in several previous
pa-pers. Gelswere treatedwithEn3Hance(NewEngland
on November 10, 2019 by guest
http://jvi.asm.org/
593
Nuclear Corp.) and dried with vacuum at60°C, and
radioactive bands were localized by autoradiography
withKodak XRP film. Exposure was for 3 to 5 days at
-700C.
Immunoprecipitation of in vitro translation products
wasperformed as described previously (13). One half
of the RNase-treated translation product (14 ,u) was
diluted with an equal volume of 2x lysis buffer and
incubated with 4 ,ul of polyclonal NC-2 serum, a
polyvalent antibody to the HSV-1 50,000-d capsid
protein (VP19C), or with 1 ,ul of Ql, a monoclonal
antibody made against purified HSV-2 alkaline
exonu-clease. Lysis buffer is 20 mM Tris-hydrochloride (pH
7.4)-S50 mMNaCl-10 mM methionine-0.5% Nonidet P-40-0.5% sodium desoxycholate-0.1% SDS. After 1
h onice, 50 ,ul of a10%suspension of Pro-ASepharose
beads (Pharmacia) in 50 mM Tris-hydrochloride (pH
7.5)-150mMNaCl-5mM disodiumEDTA-0.2%
sodi-um azide wasadded, and the suspension was
incubat-ed for a further 30 min on ice with frequent mixing.
ThePro-ASepharose beads with the immune complex
absorbedwerethendeposited by1 minof
centrifuga-tion in an Eppendorfmicrocentrifuge, and the pellet
wassuspended in 200p.loflysisbuffer and centrifuged
for 5 minthrougha1-mlpad oflysis buffer containing 1
M sucrose. TheSepharose was then washed by
sus-pension in 0.5 ml of 10 mM Tris-hydrochloride (pH
7.4)-150mMNaCI-10mMmethionine-0.2% Nonidet
P40-0.1%SDS and recentrifugation. After five wash-es, the Sepharosepellet wassuspended in 40 p.lofa
buffer containing 0.075 M Tris-hydrochloride (pH
6.8)-3% SDS-10% P-mercaptoethanol. The
suspen-sion was heated to 950C for 2 min, and the Pro-A
Sepharose was pelleted bycentrifugation. After this the supernatant, containing released immunoglobulin
andanytranslation product with which it had reacted,
wasloaded onto SDS-acrylamide gels for size
fraction-ation.
Inonesetofexperiments, we used washed,
Formal-in-killed Staphylococcus aureus (IgGsorb, The En-zymeCenter, Inc., Boston, Mass.) toprecipitate the immune complex. This method resulted in a
considera-blylargeramountofproteinprecipitatedbuta much
higherbackground of nonspecific radioactive proteins.
This procedure, however, yielded interpretable
re-sults.
Preparation of radioactive protein markers.
["S]methionine-labeled,
cytoplasmic infected-cellprotein was isolated by the basic procedure of
Ed-wards and Fan (9). Cultures (6 x 106cells) of HeLa
cellswereincubated from6 to8 hpostinfection with
200 p.Ci of
[lS]methionine
(400 Ci/mmol; NewEn-gland NuclearCorp.) in4 mlofmethionine-free Eagle
minimal essential medium.
In vitro 125I labeling of purified HSV-2 alkaline
exonuclease was carriedoutby themethodof
Green-wood et al. (14). Samples of the purified protein (0.05
p.g)werelabeled with carrier-free1251(17Ci/ml; New
England NuclearCorp.).
Preparationandcharacterizationofmonoclonal
anti-bodies against HSV alkaline exonuclease. Details of
monoclonalantibody preparation are described in the
paper by Banks et al. (in press). Briefly, HSV-2
enzymewaspurified from high-salt extracts of
HSV-2-infected Vero cells (28). Steps included
DEAE-cellu-lose, phosphocellulose, and DNA-cellulose
chroma-tography. Enzyme activity was measured by the
method of Purifoy and Powell (30). Purity can be
assessed from the data in Fig. 3A. .
Monoclonal antibodies werepreparedby two
inocu-lationsofpurified enzyme (2 to 3 p.g) into the footpads
ofBALB/c mice during a 2-week interval. Cell fusion
was by the method of Kennett et al. (17).Hybrid cell
colonies were grownin microtiter plates (Linbro) and
tested for production of antibody with an
enzyme-linkedimmunosorbent assay (horseradish
peroxidase-conjugated anti-mouse immunoglobulin G; DAKO,
Denmark) using detergent extracts of HSV-2-infected
cells as antigen. Peroxidase assay was done with
0.01% orthophenylenediamine and 0.003% hydrogen
peroxide.
Theantibody used in this paper(Q1)wasshown to
be able toimmunoprecipitate and neutralize the
HSV-2alkaline exonuclease.Furthermore, it has been used
in an immunosorbent column topurifythe enzyme. It
is strongly cross-reactive with the HSV-1-induced
enzyme (K.Powell, unpublisheddata).
RESULTS
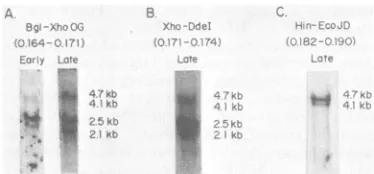
Four partially colinear mRNA species map between 0.16 and 0.19 on the HSV-1 genome. We used a number ofcDNA probes made to small (1.0- to 1.5-kilobase [kb]) fragments ofHSV-1 DNA mappingbetween 0.16 and 0.19 map units todetermine the number and time of appearance of abundant mRNAs. Four readily detectable mRNA species of approximately 2.1, 2.5, 4.1, and4.7 kb were seen inNorthern (RNA) blots of HSV-1 mRNAisolated from cells 6 h postinfec-tion. Wecould inferthattheactual mRNA sizes were 1.9, 2.3, 3.9, and 4.5 kb, since the average poly(A) tail length ofHSV-1 mRNA is ca. 200 bases(32). Nick-translated DNAprobesmapping betweenca. 0.16 and 0.174 map units hybridized to all mRNA species. Probes containing DNA mapping betweenca. 0.174 and 0.19 hybridized to only the three larger species. Examples of suchexperiments are shown in Fig. 1.
ANorthern blotof viral mRNA isolated from cells in which DNA synthesis was inhibited is
shown in Fig. 1A. Here the RNA blot was hybridized with32P-labeledDNA madeby nick-translating cloned BgIII-XhoI fragment O-G (0.164-0.171 mapunits).The2.3-kbmRNA was a majorband early, whereas the 1.9-, 3.9-, and 4.5-kbmRNAs were presentinlow abundance. The same probe revealed the 2.3-kb mRNA in equivalent abundance in Northernblots of late viral mRNA(Fig. IA). In this RNA, however, the other species were present in relatively greater abundance. This relativeabundancy dif-ference with time after infection suggested that the2.3-kbmRNAis an early
(p)
mRNA, where-asthe1.9-, 3.9-,and 4.5-kb speciesarelate(P,y) species. Criteria for temporal classification of specific mRNA specieshave been described in several recent reviews (31, 35, 36).Weused two nick-translated probes to deter-mine how far the 1.9-kb mRNA extended
be-48, 1983
on November 10, 2019 by guest
http://jvi.asm.org/
mRNAsare
considerably
lessabundant than thetwo smaller mRNAs.
E:zrl
t.+
e
High-resolution
localization of the 5' and 3'endsof the overlapping mRNA species. We used 5- and3'-end-labeled single-stranded DNAs as
k*
;r5kr
1(lhybridization
probes for precise S1 andexonu-clease VII mapping of the four mRNAslocated in Fig. 1. Basic methods have been reviewed recently(35) and are outlined above. These data
FIG. 1. In situ RNA
(Northern)
blots of HSV-1 are summarized in Fig. 2.mRNA encodedby DNA mapping between map units The 2.85-kb
BamHI-Xhol
fragment A-G 0.164 and0.190.Samples of HSV poly(A) mRNA from (0.152-0.171mapunits) was 3' end labeled at the cellsinwhich DNAsynthesis was inhibited (early) or XhoI site at map unit 0.171. Strand-separated allowedtoproceed (late) were fractionated on methyl- probe was hybridized with viral mRNA, the mercury-containing agarose gels and immobilized by hybrids were digested with S1 nuclease, and drying in vacuo. The region-specific RNA was detect- protected DNA was fractionated on a denaturingedbyhybridization with nick-translated DNA probes
acrylamide gel.
Themajor protected
species
wasas indicated. Sizes shown were determined by the
position of HeLa cell rRNA markers (not shown) as 1,600 bases long (Fig. 2A, trackS),
indlcating
a describedpreviously (1, 6). (A) mRNA species hybrid- major mRNA 3' end 1,600 bases to the left of the izing to a cDNA probe fromBglII-XhoIfragment O-G XhoI site at map unit 0.171. Minor amounts of (0.164-0.171 map units) (B) mRNA specieshybridiz-
other bands were seen with long exposures. ing to a probe from the 490 bases between theXhoI However, none contained more than 1 to 2% ofsite at 0.171 andaDdeI siteindicated in Fig. 3 and4 the total radioactivity seen. Furthermore,
simi-(below). (C) mRNA species hybridizing to a probe lar experiments (not shown) using DNA 3'-made from HindIII-EcoRI fragment J-D (0.182-0.190 labeled at the BglII site at map unit 0.163 gave a
map units). major S1 protected band 400 bases long. These
data, whentaken with experimentsbelow, indi-catedthat thefour mRNAs shareacoterminal3'
yond
the XhoI site at map unit 0.171. A probe end ca. 400bases tothe leftof theBglII site atextending
toaDdeI site490 bases to theright of 0.163. Wedidnotexcludethe presenceofsmall theXhoI site(seesequencedatabelow) hybrid- amounts of other RNA species terminating at izedtoallfourmRNAspecies(Fig.IB).
A DNA othersites.probe extending
from this DdeI siterightward
to The 5' ends of the 1.9- and 2.3-kb mRNA theHindIII siteat0.182 didnothybridizedetect-species
werelocatedby hybridizing viralmRNAably
with the 1.9-kb mRNA(not shown). with strand-separated XhoI-HindIII fragmentThe two larger
P-y
mRNAs werereadily
C'-IO DNA (0.171-0.182 map units) 5' end la-showntoextendbeyond
theHindIII siteatmap beledattheXhoI siteatmapunit0.171. Exonu-unit 0.182 since a32P-labeled
DNA probe made cleaseVIIdigestion
ofhybrids (Fig. 2B, track X) fromHindIII-EcoRI fragmentJ-D (0.182-0.190 gave three protected species. One was 1,650 mapunits) hybridized
tothem inNorthernblots baseslong, corresponding
to the full length of of late mRNA(Fig.
1C). As mentionedabove,
theDNA. Thiswas dueto theprotection
ofthe otherprobes
were used to show theprecise
3.9- and 4.5-kbmRNAs(seebelow).
Thesecondextent ofthese mRNAs. Heavier exposures of protected
fragment
was700baseslong,suggest-Northern blots hybridized with HindIII-EcoRI
ing
that the 2.3-kbmRNAextends700 bases tofragment
J-D (0.182-0.190 mapunits)
also re- theright
of the XhoI site at 0.171. The third vealed a relatively low-abundancy mRNA ca. species was 270 to 280 bases inlength.
This 2.7kb in size. This mRNA extendedtotheright
suggested that the 5' end of the contiguous oftheareaof interestpresentedinthispaper and portion ofthe 1.9-kb mRNA extendsthisfartois not characterized here. the
right
of the XhoI site. In the exonuclease VII The dataofthenextsectiondemonstrated thatdigestion
trackofFig.
2B (track X), someother the 3' end ofthefourmRNAspecies
liestothe(faint)
intermediate-sized bands were seen.left of the
BgIII
site at map unit 0.164. There- However,thesewere notconsistentlypresent infore,
theBglII-XhoI fragment
O-G (0.164-0.171 several repeatexperiments.
We suggest these map units) DNA probe could be expected to weredigestion
intermediates of the nuclease hybridizewith thefourmRNAscolinear through reaction.Si
nuclease digestion of hybrids be-thatregion
in amounts reflecting their relative tween viral mRNA andstrand-separated
XhoI-abundance. The data of Fig. 1A, then, indicateHindIll
fragment
C'-IO DNA (0.171-0.182map that the 2.3-kb(p)
mRNAis the most abundant units)5' endlabeled at theXhoI siteyieldedthe mRNA in thisregion,whereas the1.9-kbmRNA samethreefragments (Fig.2B,
track S). These is somewhat less so and the 3.9- and 4.5-kb mapping data,theclosecorrelation ofthesizeofon November 10, 2019 by guest
http://jvi.asm.org/
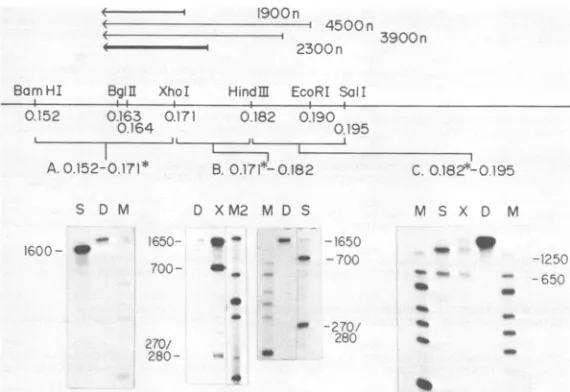
[image:4.490.54.241.71.158.2]1900 n
4500n 3900 n
2300n
Bam HI BglI XhoI HindM EcoRI Sol!
-i
0.152 0.163 0.171 0.182 0.190
0.164 0195
II A. .. .. j . In
A.0.152-0.171* B. 0.17i*-D0182
S D M D XM2 M D S M S X D M
1600-3 _167V,_.,.;50- _ 165C
40 -- 70Cr' ---'250-2K
--6r,--- ~2?C
-287 _
28zt5U f
270-280
-FIG. 2. Localization of the mRNAs by Si nuclease and exonuclease VII mapping. Single-strandedDNA5'or
3'labeledatspecific restriction siteswashybridized with HSV poly(A) mRNA, and the DNA protected from
nuclease digestion was size fractionated ondenaturing acrylamide gels asdescribed previously (13, 15). (A)
BamHI-XhoI fragment A-G DNA (0.1524.171 mapunits) 3'labeledatthe XhoI site.S, S1 nuclease-resistant
material, D,Undigested DNA,M,Size marker of Hinfi-digested pBR322 DNA; sizesare(in nucleotides from
bottom): 220/221, 298, 345, 396, 506/517, 600, and 999. (B) XhoI-HindIII fragment C'-IO DNA (0.1714.182map
units) 5' labeledattheXhoI site. D, Undigested DNA. X, Exonuclease VII-digested material. M2, Marker of
HaeII-digested pBR322 DNA; sizesare(in nucleotides frombottom): 227, 280 (partial digestion fragment), 370,
430/439, 622,and1876. S, S1 nuclease-digested material. M, Sizemarkersasdescribed for(A). (C) HindIII-SaII
fragment J-DDNA(0.1824.195mapunits) 5' labeledatthe HindlIl site.M,Size markerasdescribed for(A);S,
S1-digested material; X, exonuclease VII-digested material; D, undigestedDNA.
the Si-resistant hybrid (ca. 1,880 nucleotides) compared with the total size of the mRNA (2to
2.1 kb), and the lack of hybridization of DNA probestotheright of the DdeI sitetothe 1.9-kb mRNAall stronglysuggestthat this mRNA has
a distinct 5' end. Certainly any intron must be less than50 bases longin total length and must
be unableto hybridize efficientlywiththeDNA
underourstandard conditions.
The 5' endsof the generally colinear 3.9- and 4.5-kb mRNAs were located by usingas a
hy-bridization probe strand-separated HindIII-SalI fragment J-D (0.182-0.195 map units) 5' end labeled at the HindIII site at map unit 0.182. Here late viral mRNAprotected fragments 650 and1,250 baseslong from both S1 nuclease and exonuclease VIIdigestion (Fig. 2C, tracks S and X). Thus, the 5' ends of these mRNAsmap650
and1,250 basestotheright of the HindIlI siteat mapunit0.182. The fact that the ratio of
radioac-tivities in the two bands is similar with both nucleasedigestion regimens suggested that each mRNA has itsowndiscrete 5' end.
Furthercharacterization of the regions
contain-ing the 5' end ofthe 2.3-kb ,B and 1.9-kb I-y
mRNAs.The 1,000-base nucleotidesequence of
the DNAfromca.30bases 3' of the XhoI siteat map unit 0.171 tojust beyond the AvaI site at
0.177 is shown in Fig. 3. A summary of the results of the following experiments and the
sequencestrategy areshown in Fig. 4.
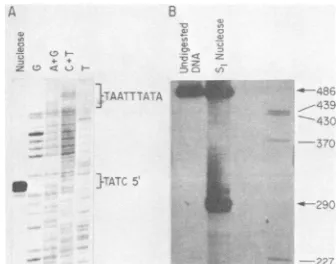
Weprecisely located the 5' end of the 2.3-kb mRNA by doing nuclease protection
experi-ments and fractionating the mRNA-protected DNA fragmenton a DNA sequencing gel. This
procedure has been documented in several
pre-vious publications (8, 12, 13). It was done by
hybridizing HSV poly(A) mRNA with single-stranded DNA, spanning the twoHinfl sites at
nucleotides 625to780, whichwas5' end labeled at nucleotide 625. Hybrids were digested with mungbeannuclease. Protected DNA was frac-tionated against a sequence ladder ofDNA 5'
labeled at the same Hinfl site (Fig. SA). Note
that the sequence ladder is of the DNA strand
complementarytothe mRNA. The5'endof the
mRNA fell within the sequence GTATC, and
thus the mRNA begins within the region be-tweennucleotides 273 and 277.
The 5'end of this mRNA fellca.28to30bases
3' tothe sequence TATAAATTA, anexcellent
TATA box. The 120or so bases5' upstream of
this 5'end share several features withpromoters
for other HSV-1 P mRNAs (reviewed in
refer-ence 35). These include a TATA box about 30
bases upstream of the transcript and an AC
C.0.182*-0195
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.490.105.390.71.267.2]1 0 20 30 40 50 60 70 80
CCGCGAGTCCACCATACCCGCCTCCTCGAGGACCACGGCCAGGGAACACAGATiTCCAGGCGGGCCCAGAGGGGACCGA GGCGCTCAGGTGGTATGGGCGGAGGAGCTCCTGGTGCCGGTCCCTTGTGTCTATTAGGTCCGCCCGGGTCTCCCCTGGCT
Xhol
90 100 110 120 130 140 150 160
TGGCCACAGGGGCGCGGACGCCGCGCAGCAACCCGCGCAGGTGGCGCTCGAACGTCTCGGCTAGTATATGGGAGGGCAGC
ACCGGTCTCCCCGCGCCTGCGGCGCGTCGTTGGGCGCGTCCACCGCGAGCTTGCAGAGCCGATCATATACCCTCCCGTCG
170 180 190 200 210 220 230 240
GCGTTGGGGATCACCGACGCCGACCACATAGAGTCAAGGTCCGGGGAGTCGGGATCGGCGTCCGGGTCGCGGGCGTGGGT CGCAACCCCTAGTGGCTGCGGCTGGTGTATCTCAGTTCCAGGCCCCTCAGC:CCTAGCCCGCAGGCCC:GCGCCCGCACCCA
250 260 270 280 290 300 310 320
GCCCCCAGGAGATAGCGGAATGTCCGGGGTCGGAGGCCCGGAGGCGTCACAAAGTGCCGGCCGACGCGGCCCGGGGCTTTT CGGGGCGTC.CTCTATCGCCTTACAGGCCCCAGCCTCCGGG.CCTCCGCAGTCTTTCACGGCCGCTGCGCCgGGCCCCGAAA
5 -1.9kb*'
330 340 350 360 370 3C0 390 400
CGTCTGCGGTGTCGGTGGCGTGCTGATCACGTGGGGGGTTAACGGGC GA6iTGGGAGCTCGGGTCCAChGCTGACCGTCGTC
GCAGACGCCACAGCCAC CGCACGACTAGTGCACCCCCCAATTGCCCGCTTACCCTCGAGC:CCAGGTGTC GECTGC'AGCAG Aval
410 420 430 440 450 460 470 480
TG0GGG0TGGGGGGGCAGGGGAC0GGAAGGTGGTTGTCAGCGCAAGACTGTTG3GGCCGGGGGCGCTTGGGGGGGCTGTCGGG
ACCCCACCCCCCCCGTCCCCTGCCTTCCACCAACAGTCGCCTTCTGACAATCCCGCCCCCGCGAACCCCCCCGACAGCCC
490 500 510 520 530C 540 550 560
GCCACGAGGGGTGTCCTCGGCCAGGGCCCAGGAACGCTTAGTCACGGTCCOTCCCGG3GGACATGCTGGW3C-CTCCCGTG G
COGGTGCTCCCC:ACAGGAGCCGGTCCCGGGTC.CTTGCGAATCAGTGCCACGC.AGGGCC.TCCTGTCCGACCCCGGAGGGCACC
Ddel
570 580 590 600 610 620 630 640
ACTCCATTTCCGAGACGACGTGGGGGA0GC GGTGGTTGAGCGCGCCGCCGGGTGAACGCTGATTCTCACGACAGCGCGTG TGAGGTAA.GGCTCTGCTGCACCCCCCTCGCCACCAACTCGCGCGGCGGCCCACTTGCGACTAAGAGTGCTGTCGCGCAC
:4- Hinf I
650 660 670 680 690 700 710 720
CCGC:GCGCACGGGTTGGTGTGACACAGGCGGGACACCAGCACCAGGAGAGGCTTAAGCTCGGG3AGGC6GCGCCACCGACG
GGCGCGCGTGCCCAACCACACTGTGTCCGCCCTGTGGTCGTGGTCCTCTCCGAATTCGAGCCCTC CGTTCCGGTGGCTGC Ava I
730 740 750 760 770 780 790 800
ACAGTATCGCCTTGTGT6TGTGCTGGTAATTTATACACCGATCCGTAA3C GCGCGCCGAATCTTGGCiATTGCGGAGGTGG TGTCATAGCGGAACACACACACGACCATTAAATATGTGGCTAGGCATTTGCGCGCGGCTTA0AACCCTAACGCCTCCACC
°5- 2.3kb HinfI
810 020 830 840 850 860 870 880
CGCCGGATGCCCTCTGGGACGTCATACGCCAGGCCGTGGGTGTTGGTCTCGGCCGAGTTGACAAACAGGGCTGGGTGCAG GCGGCCTACGGGAGACCCTGCAGTATGCGGTCCGGCACCCACAACCAGAGCCGGCTCAACTGTTTGTCCCGACCCACGTC
890 900 910 920 930 940 950 960
CACGCAGCGATAGCGCAGCAGGGCCAGGGCGAAGTCC0TCGACAGCTGGTGTTTT0ATACTGGTAACCGG CCGGG GTGCGTCGCTATCGCGTCGTCCCGGTCCCGCTTCAGGCAGCTGTCGACCAACAACTTTATGACCATTGGCCCTTTGOCCC
970 980 990 1000
TCACGGGTACGCCCAGGCTCGGGGCOACGTACACGCTAAC AGTGCCCATGCGGGTCCGAGCCCCGCTGCATGTGCGATTG
FIG. 3. Nucleotide sequence ofDNA betweentheXhoIsite at map unit 0.171 and the AvaIsite at 0.177.
Sequencing
wasbythe method of Maxam andGilbert(23), and the strategy is shown in Fig. 4. The locations ofthe 2.3- and 1.9-kb mRNA5'ends
(*-O),
in-phase translation initiators( 3),and several other features of thesequenceareindicated.
0 (100) 200 300 400 500 600 700 800 900 1000
Hinf I AvaI DdeI AvaI Pvull
Xhol HinfI Smal Pvull HinflHinfl Hinfl AvaI
-, -....
-2.3
kbJ13)
mRNA 3.9 kb(13)1
mRNA 4.5kb(IY)
mRNA1.9kb
(or)1
mRNA Translation Frames-3 -3
-3-
9:43
IK 1FIG. 4. Sequencing strategyand summary. High-resolution restriction maps forXhol, Hinfl,
Sinal,
Aval, Pvull,andDdel sites areshown, aswellasthe lengthofsequence ladders from DNA 5' labeledat suchsitesThelocations ofthe 5' ends of the 2.3-kb(p)and the 1.9 kb
(P3y)
mRNAsand of thetranslationalstartE)andstop (Z)codonsare shown.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.490.106.391.69.376.2] [image:6.490.110.391.470.625.2]HSV mRNA 597
S~~~~~~~~~~~~~~~~~~~~~~~~~~L , .._
.... 3D. #;.
-_
.
FIG. 5. (A)Precise localization of the
2.3-kb alkaline exonuclease mRNA. I
mRNA washybridized with single-strar
end labeledattheHinfl siteatnucleotid
andextendingtotheHinfl siteatnucleo
was then digested with mung beannucli
protected DNA wasfractionated agains
ladderof DNA 5' labeledatnucleotide 62
localization of the 5' end of the 1.9-kbr
spanning from the XhoI siteatnucleotide
the Ddel siteatnucleotide 520 was 5'e
base 28 and strandseparated. Itwashy
viral mRNA, thehybridsweredigestedv
ase, andprotected DNAwasfractionati
turingacrylamide gel, usingHaeII-cut I
fragments as asize standard.
string110 to 120bases upstream. T son of the 120 bases upstream o characterized ,B mRNAsis shownin comparison gives us reasonable cc
concluding
thatthis regionupstream4of the2.3-kb mRNA is, indeed, a, ]
-120
EARLY (BETA) HSV PROMOTORS
-90
The sequence data in Fig. 3wereanalyzedfor potential translational initiation and termination codons. The 5' end of the 2.3-kb (1) mRNA is 160bases upstream of the canonical eucaryotic translation initiation sequence AAATGG (nucle-otides 432 to 437), as described by Kozak (18). This translation frame (frame 1 of Fig. 4) stays openthroughout the region sequenced and con-tainsmethionine codons 379, 553, and 643 bases downstream. It is a reasonable assumption that this frame defines the N-terminal region of a protein. A second ATG codon isseeninreading frame 3 at position 457-459, but this frame is terminated24 bases downstream with the TAA 5' end of the triplet atposition 481-483.
HSV poly(A) All reading frames contain multiple
termina-nded
DNA 5 tors upstream ofthepotential translation frame le622(Fig. 3) for the 2.3-kbmRNA, so anyprotein encoded bylease
andthe
the 3.9- or4.5-kb mRNAs would notshare anyst
asequence
translational frames with aprotein
encodedby
2.(B) Precise the 2.3-kb mRNA.
mRNA. DNA We hybridized single-stranded DNA, 5' end 28(Fig. 3) to labeled at the XhoI site (base 28; Fig. 3) and nd labeled at extending to the DdeI site at base 520, with viral 'bridized with mRNA and
carried
out Si nuclease analysis.with
S1
nucle- Twoprotected specieswere seen. One migrateded on adena- with asize of 290 bases, and one migrated along
pBR322
DNA with undigested DNA (Fig.SB).
The shorter DNA species corresponded to the 270- to 280-base bandseenin Fig.2B (tracks S andX). The full-length band indicated that no other major 'he compari- species was seen in the region extending up tof five well- 520 basesupstream of the XhoI site at 0.271. ^Fig.6.This Control experiments (not shown) indicated )nfidence in that S1 nuclease-protected DNA was entirely of the 5' end dependent on the presence of viral mRNA in the promoter. hybridization. We precisely located the mRNA
-60 -30
Alkaline Exonuclease (2.5kb)
Transcribed from rightto left with 5' end at 0.175
AGACCAACACCCACGGCCTGGCGTATGACG TCCCAGAGGGCATCCGGTGCCACCTCCGCA ATCCCAAGATTCGGCGCGCGTTTACGGATC GGTGTATAAATTACCAGCACACACACAAGA
Thymidine kinase-type 1 (1.5 kb)
Transcribedright toleft with5' end at 0.315
CTATGATGACACAAACCCCGCCCAGCGTCT TGTCATTGGCGAATTCGAACAGGCAGATGC AGTCGGGGCGGCGCGGTCCGAGGTCCACTT CGCATATTAAGGTGACGCGTGTGGCCTCGA
5.2 kb mRNAencodingHSV-1 140k protein Transcribed left to rightwith 5' end at0.565
CATGGAAGGAACACACCCCCGTGACTCAGG ACATCGGCGTGTCCTTTTGGGTTTCACTGA AACTGGTCCGCGCCCCACCGCTGCGCGATG TGGATAAAAAGCCAGCGCGGGTGGTTTGGG
1.5 kbmRNAencoding HSV-1 38k protein
Transcribed left torightwith 5' endat0.590 CGACCATAGCCAATCCATGACCCTGTATG TCACGGAGAAGGCGGACGGGACCCTCCCAG
1.5kb mRNA encoding 39k protein
Transcribed left to rightwith 5' end at 0.699
;CATATAAGCGCGGACTAAAAACAGGGATGT
CCC CTCACCCCACACAGGGCGGGTTCAG GCCTGCCCGGCAGCCAGTAGCCTCTGGCAGiATCTGACAGAC(iTGTGCGATAATACACACG CCCATCGAGGCCATGCCTACATAAAAGGGC
FIG. 6. Sequence comparison for the promoters of HSV-1 ,B mRNAs. The 2.3-kb mRNA promoter region
from the data of Fig. 3 is shown compared with data for other HSV-11 mRNAs. The data for HSV-1 thymidine
kinasearefrom McKnight (24), those for the 5.2-kb mRNA are fromFrink et al. (13), those for the 1.2-kb mRNA
arefrom Draper et al. (8), and those for the 1.3-kb mRNA are from Hall et al. (15).
VOL.48, 1983
7
c-.
,,:-'L
,.4--r - ;%
I " "
i
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.490.61.229.73.205.2]598
end (seen above) to around base 320 by using high-resolution sequencing gels against se-quence ladders (not shown). This putative cap site does not lie 25 to 30 bases downstream of any exceptionally appealing TATA box se-quence. However, the sequence between bases 350 and 345 (TGATC) could conceivably serve as an equivalent feature. The cap site does lie 131 bases to the right of the ATG triplet at position 187-189. This could serve as a transla-tional initiator. If so, the polypeptide encoded should share the same reading frame as that encoded by the 2.3-kb mRNA.
Evidence that the 2.3-kb earlymRNA encodes
alkalineexonuclease. We used a monoclonal anti-body against HSV-2 alkaline exonuclease to demonstratethat the 2.3-kb mRNA encodes the cross-reactive HSV-1 protein. Denaturing gel
electrophoresis showed that this antibody (Q1) specifically binds to purified '251-labeled HSV-2
alkaline exonuclease (82,000 molecular weight) (Fig. 7, track Q1). Another monoclonal antibody (T2/Ti), which is directed against other specific
sitesonthe HSV-2 enzyme, did not precipitate it
(Fig. 7A, track
T2/T1).
This demonstrated the specificity of the Ql precipitation. Ql reactedwith the 82,000-d HSV-1 enzyme from
[35S]methionine-labeled, HSV-1-infected cell extracts (Fig. 7A, track Ql-I.P.). Other faint protein bands were also visualized. No specific protein band could be seen when Ql was reacted with uninfected cell extract (data not shown). Therefore, it is not known whether these
repre-sent nonspecific binding or binding of antigeni-cally related, infected-cell proteins orboth.
Wepurified mRNA encoded in this region of HSV-1 DNA by using BamHI-HindIll fragment A-IO (0.152-0.182 map units) and XhoI-HindIII fragment C'-IO (0.171-0.182 map units) bound
to cellulose. Both fragments hybridized to all mRNAs (see Fig. 1), but the former fragment also encodes the 3' region of another 2-kb mRNAwhose 5' end maps near 0.147 and whose 3' end maps near 0.160 (data not shown). We subjected a sample of purified mRNA (ca. 0.1 p.g
oftotal mRNA) to in vitro translation by using
[35S]methionine
and incubated the total transla-tion mix with Ql. Reactive protein was size fractionated on a denaturing SDS-acrylamide gel (Fig. 7B, tracks C'-IO and A-IO). Both mRNA preparations translated the same 82,000-d poly-peptide. In vitro translation of HSV-1 mRNA purified by using DNA fragments from other regions of the genome did not yield any detect-able protein reactive withQi
antibody (data not shown).Inanother experiment,wepurified mRNA by usingXhoI-HindIIIfragment C'-IO (0.171-0.182 mapunits) andtranslated it invitro. Translation products were reacted with Ql antibody. This
FIG. 7. In vitro translation and identification of
HSV-1 alkaline exonuclease. All autoradiographs are
ofdenaturing SDS-acrylamide gels(19). Sizes of
pro-teins (X103 d) were determined by comigration with
adenovirus mRNA translation products (not always
shown) asdescribedpreviously(1). (A)Ql,
Immuno-precipitation of125I-labeled HSV-2 alkaline
exonucle-ase with monoclonal antibody Ql.
T,/Tj,
Lack ofprecipitation with monoclonal antibody TJ/T1. En-zyme, Authentic enzyme. Ql-I.P.,
Immunoprecipita-tion ofcytoplasmic, 35S-labeled, infected-cell protein
with monoclonal antibody Ql. I.P., Cytoplasmic
in-fected-cell protein. (B)Immunoreactivities of in vitro
translationproducts.Cl10-Ql,Productsoftranslation
of Xhol-Hindlll fragment C'-IO DNA (0.171-0.182
map units) specific mRNA immunoprecipitated with
Ql. AIO-Ql, The same immunoprecipitation, except
thatBamHI-HindlllfragmentA-10DNA(0.152-0.182
map units) specific mRNA was used for the in vitro
translation.Ad, In vitro translationproductsof
adeno-virusmRNA. (C) ComigrationofHSV-2 enzymeand
HSV-1 in vitro translation product. HSV, HSV-1
poly(A) mRNA (1
p.g)
in vitro translation products. Enzyme-Ql, 125I-labeled HSV-2 alkaline exonucleaseprecipitated with Ql. Cl10-Ql, In vitro translation
productofXhol-Hindlllfragment C'-4ODNA
(0.171-0.182mapunits)specificmRNAprecipitatedwithQl.
(D) Translation of size-fractionated HSV poly(A)
mRNAandprecipitation withQl. Sizes ofmRNAare
indicated above the appropriate tracks. I.p.,
35S-la-beled, cytoplasmicinfected-cell protein.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.490.256.442.98.365.2]HSV mRNA 599
material was then subjected to denaturing gel electrophoresis along withpurified, 125I-labeled HSV-2 alkaline exonuclease which had also beenimmunoprecipitatedwith Ql antibody (Fig. 7C). This experiment confirmed the essential comigration of the HSV-1 in vitro translation product with authentic HSV-2 alkaline exonu-clease.
The experiments described above established thefact that oneof the fourmRNAs located in the region between 0.160 and 0.190 map units encodes HSV-1 alkaline exonuclease. We trans-lated size-fractionated total HSV-1 mRNA and used Ql antibodytoobtainaprecise size for the mRNAencoding theenzyme. We size fraction-ated 10,ugof HSV poly(A)mRNA on a denatur-ingagarose gel. Four mRNA pools were taken, migrating with sizes between 5.2 and 4 kb, 3.7 and 3.1 kb, 2.9 and 2.5 kb, and 2.3 and 2 kb. Each was eluted, reselected on oligodeoxythy-midylic acid-cellulose, and subjected to in vitro translation. The translation products were then reactedwith Ql antibody. It was clear(Fig.7D) that major synthesis of the 82,000-d alkaline exonuclease is mediated by mRNA in the size range of2.9 to 2.5 kb. These data demonstrate thatthe 2.3-kb ,B mRNA does, indeed, encode the enzyme. [Note: as in Fig. 1, this mRNA migrates with a rate corresponding to a size of 2.5kbbecause of the poly(A) tail.] The radioac-tivity migratingat-50,000 d in each track is an endogenousband from the reticulocyte system. Its presence here is duetothefact thatweused IgGsorb instead of Pro-A Sepharose to react
with the immune complex because of the
former'sgreaterabilitytobindtoimmune com-plexes.
Identificationof a potential translation product forthe 1.9-kb mRNA.Partialsequencedata(Fig. 3)indicated that the 1.9-kbmRNAcolinear with the 3'region ofthe2.3-kbmRNAcould encodea polypeptide about 130 amino acids shorter than
the alkaline exonuclease. We used
BamHI-HindIlI fragment
A-TO
(0.152-0.182 mapunits) bound to cellulose to isolate all the mRNAs encoded in the region of interest. We size frac-tionatedtheseregion-specificmRNAs and trans-lated both the 2.3-kb and 1.9-kb mRNAs in vitro. We used methods described in detail in several recentpublications (see reference 13) for the size fractionation of the hybrid-selected mRNA.Translation of the 2.3-kb mRNA yielded anumberofpolypeptides, butonly the 82,000-d alkaline exonuclease was reactive with Ql anti-body (Fig. 8,tracks T-1 and Q1-1).The mRNA isolated as the 1.9-kb species actuallycontains both this mRNAand a 1.9-kb mRNA whichis homologous to the other DNA strand and has a 3' region homologous to the region between 0.152 and 0.160 map units (see
P'>
mF;;-w23bc- r
.s _
z
f7 A
"._
-.
FIG. 8. In vitro translation of the 1.9-kb mRNA.
The 2.3-kb and 1.9-kb hybrids, selected by using
BamHI-HindIII fragment A-IO (0.152-0.182 map units) bound to cellulose, were fractionated by
electro-phoresis on a denaturing agarose gelcontaining
meth-ylmercury hydroxide. Each was translated in vitro;
halfof each translation product was fractionated
with-outfurther treatment (tracks T), andhalf was reacted
with Ql monoclonal antibody (tracks Q1). Details of the translation and fractionation of products are as
described for Fig. 7. A track of the translationproduct
of rabbit globin mRNA is includedas acontrol.
the preceding section). Translation of this mRNA mixture consistently gave two polypep-tide products distinct from endogenous bands found in the translation system. The larger mi-grated witha rate
corresponding
toamolecular weight of60,000, and the smallermigratedwitharate correspondingto54,000(Fig. 8,trackT-2). We suggest that oneof these
polypeptides
is the translationproduct of the 1.9-kbmRNA in ques-tion. Since, however, neither product reacted withQl antibody (Fig.8,trackQ1-2),our identi-fication of a translation product for the 1.9-kb mRNAdescribed in thisreportisonlytentative. Tentative evidence that a50,000-d HSV-1 cap-sidprotein is encoded by either the 3.9- or 4.5-kb mRNA. We used apolyclonal rabbit antiserum made against the SDS gel-purified 50,000-d HSV-1 capsid protein (VP19C) to tentatively identify itas atranslationproduct of the 3.9- or 4.5-kb mRNA.This antiserum(NC-2) specifical-lyreactswitha50,000-d protein from [35S]meth-ionine-labeled extracts from HSV-1-infected cells. Adenaturingacrylamide gelsize fraction-ationofinfected-cell proteinreactive with NC-2 is shown in Fig. 9A (track NC2-I.P.). Some other proteins are also detectable, especially with long exposures (ca. 2 weeks). Aprominent oneis the155,000-dcapsid protein, ICP5,which may have been a contaminant of the original size-fractionated protein antigen.We prepared purified
region-specific
viralmRNA, using BamHI-HindIII fragment A-TO
VOL.48, 1983
.-',i
el"..,"
a o.,;,tD.1..-.-n!'il. -
-.4
.:7, 4-ll.- -j
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.490.289.410.65.220.2]FIG. 9. Immunoprecipitationofa50,000-d protein with antiserum NC-2. Details are asfor Fig. 7. (A)
Comigration ofproteinprecipitablefromcytoplasmic
infected-cell proteinand in vitro translation product.
I.P., 35S-labeled cytoplasmic infected-cell protein.
NC2-I.P., Cytoplasmicinfected-cellprotein
precipitat-ed withNC-2 antiserum. NC2-A-IO, In vitro
transla-tion products of BamHI-HindIIIfragmentA-IO DNA
(0.152-0.182 map units) specific mRNA precipitated
with NC-2 antiserum. (B) Antiserum NC-2
precipita-tionofa50,000-dtranslation productofHindIII-SaIl
fragment J-D (0.182-0.195mapunits) specificmRNA.
I.P., Cytoplasmic infected-cell protein. NC-2 No
RNA, Material precipitable with NC-2 and IgGsorb
fromthetranslationproductsresultingfromnoadded
RNA.NC-2HS-JD, Proteinsprecipitated usingNC-2
andIgGsorb fromthe translation products of
HindIll-SallfragmentJ-Dspecific mRNA. The bandmigrating
at -65,000d(?)wasnotidentified.
(0.152-0.182 map units) and XhoI-HindIII frag-mentC'-IO(0.171-0.182mapunits) DNA bound tocelluloseasdescribed in the previous section.
We used this mRNA asa template for in vitro
translation and used the NC-2 antiserum as a reagent to detect the 50,000-d capsid protein. Both fragments consistently gave positive
re-sults. However, the amount ofradioactivity in the 50,000-d protein isolated was considerably
lessthanthat in the82,000-d protein isolated by using Ql antibody against alkaline exonuclease. This conclusion was based on the fact that
autoradiographs required three to five times
moreexposuretoobtainequivalentexposuresof
the 50,000-d protein compared to the 82,000-d
one. Atypical experimentusing BamHI-HindIII
fragment A-IO (0.152-0.182mapunits) DNAfor
hybrid selection is represented in Fig. 9A (track NC2-AIO).
Thebandofradioactivitymigratingat82,000 d is alkaline exonuclease, which is also translated fromthis mixedmRNApreparation (seeabove). We could notdetermine whether this ability of NC-2 antiserum to react with this enzyme was
due to specific immunological cross-reactivity.
Long exposures of immunoprecipitates of
35S-labeled infected-cell protein extracts, such as
shown in Fig. 9, did reveal a band of protein
migrating witha rate correspondingto 82,000 d in size. However, nonimmune rabbit serum
boundpurified
251I-labeled
alkaline exonuclease unlesslarge
amounts of carrier protein werepresent. Therefore, the precipitation ofthe
en-zymeseen withtheinvitrotranslation couldbe nonspecific binding. Ql did not appear toreact
with the50,000-dproteinsynthesizedin vitroas
judged byverylongexposures(3months)of the autoradiographs seen inFig. 3.
Therelativespecificityof theNC-2
antiserum,
aswell as thecomigration of immunoprecipitat-ed infectimmunoprecipitat-ed-cell 50,000-d capsid protein and the in vitro translation product (compare Fig.
9A,
tracks NC2-I.P. and NC2-AIO), suggested that the mRNA for thisproteiniseitherthe3.9-kborthe 4.5-kb
(P3y)
mRNA mapped in Fig. 2. This identification must be regarded as tentative, however, since the amount ofradioactivity iso-lated in the 50,000-d translation product preclud-ed tryptic peptide comparison with thepurified capsid protein. The relatively small amount of protein recovered from the in vitro translation product could be due to any combination of three factors. First, total translation product contains arelatively small amount of the 50,000-dprotein, equivalent to endogenous products of translation (data not shown). This reflects the fact that neither of the two mRNAs is highly abundant. Second, the methionine content of the proteinis unknown and may be low. Third, the NC-2 antiserum was made against denatured 50,000-d capsid protein and may not react effi-ciently with protein translated in vitro.We confirmed our inference that either the 3.9-kb or4.5-kb
(B-y)
mRNAencodeda50,000-dpolypeptide in two ways. First, we used
HindIII-Sall fragment J-D (0.182-0.192 map units) DNA bound tocellulose topurifythe 3.9-kb and 4.5-kb mRNAs away from the 2.3-kb mRNA for alkaline exonuclease (see Fig. 1). Suchhybrid purifiedmRNA, when translated in vitro, yielded the 50,000-d protein immunoreac-tive with NC-2 antiserum (Fig. 4B). Here, the use of IgGsorb led to the recovery of other bands due to endogenous translation products, but thepresence of the 50,000-d protein specific for the viral mRNA is clear. Second, we used size-fractionated total HSV poly(A) mRNA to translatethisprotein. Thisexperiment(data not shown) wasdone as describedin the preceding section, except that mRNA ranging from a size of 5.0 to 4.5 kb wastranslated and then reacted with NC-2 antiserum. Here otherprotein bands were also seen, but the 50,000-d protein was clearly translated by mRNA of the size range between 5 and 4 kb. Larger and smaller size fractionsdid not translate thisprotein.
DISCUSSION
The moderate-resolution transcription map presented inFig.2isentirelyconsistent with the
VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
HSV mRNA 601
picture of HSV-1 gene expression and gene packaging developed in the last several years (reviewed in reference 35). The fact that the mRNAs characterized can be correlated with specific viral genes demonstrates, again, the genetic resolution available through HSV tran-scription mapping.Weshouldnote attheoutset, however, that none ofthe datapresented here precludes the presence of small amounts of othertranscripts in this
region
being
generated via splicing from other promoters orby use of otherpolyadenylation sites.Ourconfidence in the identification of the 2.3-kba mRNA asencoding alkaline exonuclease is quite high. The immunereactivity with the high-ly specific monoclonal antibody Ql is, itself, strong evidence, especially when migration of theinvitro translation product is compared with that ofauthentic enzyme and with that seenin the infected cell (Fig. 7). The fact that Preston andCordingley(29)also located thegenefor this important enzyme in this region adds to the alreadystrong case. It should benoted, howev-er, that these workers estimated themRNAfor the enzyme to be in the size range of 3.6 kb, considerably greaterthan that seen here. Their result could be due to the procedures used to fractionate the RNA (sucrose
gradients).
It could bedue,however,totheamphibian
oocyte system they used for translationbeing
able to initiate translation in the interior of the partially colinear 3.9-and 4.5-kb mRNAs. Basedon the present data, we conclude that these mRNAs contain all the information for the alkalineexo-nucleasedownstream of their translational read-ing frames. Such patterns of long untranslated regions ofsomelarge HSV-1 mRNAs have been described byus
previously.
Our confidence in the conclusion that the 50,000-d
capsid
protein
VP19C is encodedby
either the 3.9- or4.5-kb mRNA is not as high. Certainly, these mRNAs encode a protein mi-grating at 50,000 d, but the relatively weak reaction between the NC-2 antiserum and thein vitro translation products must indicate some caution. It is conceivable (although
unlikely)
that the
precipitation
of the translation product by this antiserum is nonspecific. The fact that the capsid protein has been mapped in this general location (20)appears to make this possi-bilityevenlesslikely.Thepartial sequence analysis presented here, and further sequence analysis currently under way in this laboratory, are useful in defining potential promoters and translational reading frames. Ourlocation of the
5'
end of the 2.3-kb alkaline exonuclease mRNA is quite solid and places thismRNAimmediately downstreamof a 120-basesequence containing features of HSV-1 1 promoters. We first noted the similaritybe-tweenanother,Bpromoter(forthe5.2-kbmRNA mapping in HindIII fragment K) and that for HSV-1 tk several years ago (12). Subsequent studies have tended to confirm thegeneral fea-turesnoted. These studies have beenreviewed, but the data of Fig. 6 are the most complete comparison available at this time.
It is clear from the analysis ofthe potential reading framessummarized in Fig.4that thereis noapparentsharedreading framefor the 82,000-dalkalineexonuclease and the 50,000-dVP19C. This isconsistent with results from other regions of the genome containing partially colinear mRNAs. We cannot be absolutely certain that translation frame1of Fig. 4 doesindeed encode the alkaline exonuclease, nor that frame 3 (which initiates further upstream) encodes VP19C. We arecertain, however,ofourfinding oftranslational stop signals in all three frames before the startsignal of frame 1. Furthermore, sequence data not shown indicate that frame 1 stays open for at least several hundred bases downstream of the data shown and that frame 3 staysopenforatleastthesame amountofbases upstream.
Given the size of 2.3 kb as the actual tran-script size for alkaline exonuclease and transla-tion frame 1 as its properframe, we can infer that the total mRNA coding capacity is on the order of2,100 nucleotides. Such would encodea protein of 700 amino acids, somewhat small for the measured size of alkaline exonuclease (82,000d). However, thereare37prolines inthe first 236 positions in the predicted amino acid sequence. Suchahigh prolinecontent(ca. 15%) would lead to a protein migrating
.more
slowly than predicted from its amino acid residue weight. We havecitedthisas the reasonforthe discrepancy between the predicted amino acid residue molecular weight of HSV-1 gC and its migration rate (60,000dversus 69,000 d;13).Partial sequence data (not presented here) around the 5'ends ofthe 3.9- and 4.5-kbmRNAs suggest that the larger of the two mRNAs en-codesashortreading frame which terminates in the leader of the 3.9-kb mRNA. Furthermore, datasuggest that the reading frame seen in the 3.9-kbmRNAis open forasignificant stretchof nucleotides. Such data indicate that the 3.9-kb mRNA,then, actually encodes the 50,000-d pro-tein that we have suggested is VP19C. This situation wouldbesimilartothatseenbyHall et al. (15) for two partially colinear and comple-mentaryoverlappingmRNAsin theregionabout map unit 0.7.
The mostdisturbingaspect of ourconclusion that the 50,000-d translation productis actually VP19C is the lowabundanceof the mRNA. We should state, however, that the amount of
[35S]methionine-labeled
VP19C frompurifiedvi-VOL.48, 1983
on November 10, 2019 by guest
http://jvi.asm.org/
rions is very much less than that of VP5 (5). Also, we have found that the total amount of VP19Crecoverablefrominfected-cellextracts is very much less than that of VP5 by using im-mune precipitation (Costa and Wagner, unpub-lisheddata). Therefore, theamountof VP19C in theinfected cell maybe low and the abundance of the mRNA
encoding
itmay also be low.A striking result in terms of HSV-1 mRNA expression ofourpresent study is the
finding
of a smaller mRNA underlying the 3' three-quar-tersof the 2.3-kbmRNAandapparentlysharing atranslational reading frame with it. Other ex-amples of shared translational reading frames arefound in the HSV-1thymidine kinasegene as shownby Marsdenetal. (22)and,potentially, in the gCgene (13).The sequence data suggest that any protein encodedby the 1.9-kbmRNAshould contain the 570C-terminal amino acids of alkaline exonucle-ase. Thisprediction is certainly consistent with the size for either in vitro translation product seen in Fig. 8. We can assume that the type-commonepitoperecognized by the Ql antibody is dependent upon the N-terminal 130 amino acids of the alkaline exonuclease. Theproperties andpossiblebiological function ofanytruncated form of the alkaline exonuclease which would lead to its being required at late times after infection are unknown. Indeed, it should be
emphasized
that such aprotein
has not been describedasbeing
presentin normallytic
infec-tionby HSV. Furthersequenceanalysis,aswellasfurthermRNAand
protein
fractionation pro-cedurescurrently
being
undertaken in ourvari-ous
laboratories,
may lead to morespecific
predictions
regarding
thisputative protein. The datadosuggest,however, that thepartialcolin-earity
ofmRNAsunderindependent temporalor promoter control can be a result of related features of theproteins encoded.ACKNOWLEDGMENTS
We thank L. Hall, M. Rice, and R. Frinkfor help and discussion.
Workwas supportedby Public Health Service grant CA-11861from the National Cancer Institute andgrantMV-159 from the American CancerSociety toE.W. and by Public
HealthServicegrantsDE-02623(NationalInstitute of Dental
Research) and AI-18289 (National Institute of Allergy and InfectiousDisease)toR.E.and G.C. R.C. isatrainee under Public Health Service Molecular and CellularBiology training
grantGM-07311.
LITERATURECITED
1. Anderson,K.P.,R.J.Frink.G.B.Devi, B. H.Gaylord,
R. H.Costa,andE.K.Wagner.1981. Detailed
character-ization of the mRNA mapping theHindlIl fragment K regionof theherpessimplexvirus type1genome.J. Virol. 37:1011-1027.
2. Anderson, K.P., J. Stringer, L. Holland, and E.Wagner. 1979. Isolation and localization ofherpes simplex virus type 1 mRNA. J. Virol. 30:805-820.
3. Bailey, J. M., and N. Davidson. 1976.Methylmercury asa reversible denaturing agent for agarose gel electrophore-sis.Anal. Biochem. 70:75-85.
4. Berk, A.J., and P. A. Sharp. 1977. Sizing and mapping of early adenovirus mRNAs by gel electrophoresis ofS1 endonuclease-digested hybrids. Cell 12:721-732. 5. Cohen, G. H., M. Ponce de Leon, H.Diggelmen, W. C.
Lawrence, S. K. Vernon, and R. J. Eisenberg. 1980. Structuralanalysis of the capsid polypeptides of herpes simplex virus types 1 and 2. J. Virol. 34:521-531. 6. Costa, R. H., B. G. Devi, K. P. Anderson, B. H. Gaylord,
and E.K. Wagner. 1981. Characterization of a major late herpes simplex virus type 1 mRNA. J. Virol. 38:483-4%. 7. Denhardt, D. T. 1966. A membrane-filter technique for the detection ofcomplementary DNA. Biochem. Biophys. Res.Commun. 23:641-646.
8. Draper, K. G., R. J. Frink, and E. K. Wagner. 1982. Detailedcharacterization of an apparently unspliced 1 herpes simplex virus type 1 gene mapping in the interior of another. J. Virol. 43:1123-1128.
9. Edwards,S.A.,andH.Fan.1979.gag-Related polypro-teins ofMoloney murine leukemia virus: evidence for independent synthesisofglycosylatedandunglycosylated forms. J. Virol. 30:551-563.
10. Franke, B., H. Moss, M. C.Timbury, and J. Hay. 1978. Alkaline DNaseactivityin cellsinfected witha tempera-ture-sensitivemutantofherpes-simplex virus type 2. J. Virol. 26:209-213.
11. Frink. R.J., K. P. Anderson, and E. K. Wagner. 1981. Herpessimplex virus type 1HindlII fragment L encodes spliced and complementary mRNA species. J. Virol. 39:559-572.
12. Frink. R. J., K. G. Draper, and E. K. Wagner. 1981. Uninfected cell polymerase efficiently transcribes early but not late herpes simplexvirus type 1 mRNA. Proc. Natl. Acad. Sci. U.S.A. 78:6139-6143.
13. Frink,R.J., R.Eisenberg,G.Cohen,andE.K.Wagner. 1983. Detailed analysis of the portion of the herpes simplexvirus type 1 genomeencoding glycoproteinC. J. Virol. 45:634-647.
14. Greenwood, F., W. Hunter, and J. Glover. 1963. The preparationof125I-labeledhumangrowthhormoneofhigh specificradioactivity.Biochem. J. 89:114-123. 15. Hall, L.M.,K.G.Draper,R.J.Frink,R.H.Costa, and
E. K.Wagner.1982.Herpes simplexvirus mRNA species mappinginEcoRIfragmentI.J. Virol. 43:594-607. 16. Hoffman, P.,andY.Cheng.1977. Thedeoxyribonuclease
induced after infection of KB cells by herpes simplex virus type1 ortype 2. J.Biol. Chem. 253:3557-3562. 17. Kennett, R. H., K. A. Denis, A. S. Tung, and N. R.
Klinman. 1978.Hybridplasmacytoma production:fusion with adultspleencells, spleenfragments,neonatalspleen cells and human spleen cells. Top. Microbiol. Immun. 81:77-91.
18. Kozak, M.1981. Possiblerole offlankingnucleotides in recognition ofthe AUG initiator codon by eukaryotic ribosomes. Nucleic Acids Res. 9:5233-5252.
19. Laemmil, U. K. 1970. Cleavage of structural proteins
during the assembly of the head ofbacteriophage T4.
Nature(London)277:680-685.
20. Lemaster, S.,andB.Roizman. 1980.Herpes simplex virus phosphoproteins. II.Characterization of the virion pro-tein kinase and ofthepolypeptidesphosphorylated inthe virion. J.Virol. 35:798-811.
21. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring HarborLaboratory,ColdSpring Harbor,N.Y. 22. Marsden, H. S., L. Haarr, and C. M. Preston. 1983.
Processingofherpessimplexvirusproteinsandevidence thattranslationofthymidinekinasemRNAis initiatedat
threeseparateAUG codons. J.Virol. 46:434-445.
23. Maxam,A.,and W.Gilbert. 1980.Sequencingend-labeled J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
DNA with base-specific chemical cleavages. Methods Enzymol. 65:499-559.
24. McKnight, S. L. 1980. The nucleotide sequence and transcript map of the herpes simplex virus thymidine kinasegene. Nucleic Acids Res. 8:5949-5964.
25. Morrison, J., and H. Keir. 1968. A new DNA exonuclease in cells infected with herpesvirus: partial purification and properties of the enzyme. J.Gen. Virol. 3:337-347. 26. Moss,H., P. Chartrand, M. C. Timbury, and J. Hay. 1979.
Mutantof herpessimplex virus type2with temperature-sensitive lesions affecting virion thermostability and DNaseactivity: identification of the lethal mutation and physical mapping of thenuc- lesion. J. Virol. 32:140-146. 27. Palmiter, R. D. 1974. Mg++ precipitation of ribonucleo-protein complexes. Expedient techniques for the isolation of undegraded polysomes and messenger ribonucleic acid. Biochemistry 13:3606-3614.
28. Powell,K. L., and D. J. M. Purifoy. 1977. Nonstructural proteinsof herpes simplex virus. I. Purification of the induced DNA polymerase. J. Virol. 24:618-626. 29. Preston,C. M., and M. G. Cordingley. 1982. mRNA- and
DNA-directed synthesis of herpes simplex virus-coded exonuclease in Xenopuslaevis oocytes. J. Virol. 43:386-394.
30. Purifoy, D.J. M.,andK. K.Powell.1976.DNA-binding
proteins inducedby herpes simplex virus type 2 inHEp-2 cells. J. Virol. 19:717-731.
31. Spear, P., and B. Roizman. 1980. Herpes simples virus, p. 615-746. In J. Tooze (ed.), Molecularbiology of tumor viruses: DNA tumor viruses, part 2, 2nd ed. ColdSpring Harbor Laboratory, Cold Spring Harbor, N.Y. 32. Stringer,J., L. Holland,R.Swanstrom, K. Pivo,andE.
Wagner.1977.Quantitation of herpes simplex virustype 1
RNA in infected HeLa cells. J. Virol. 21:889-901. 33. Strobel-Fidler, M.,and B. Franke.1980. Alkaline
deoxyri-bonuclease induced by HSV type 1: composition and properties of the purified enzyme. Virology 103:493-501. 34. Vernon, S. K., M. Ponce de Leon, G. H.Cohen, R. J. Eisenberg, and B. A. Rubin. 1981. Morphological
compo-nentsof herpesvirus. III. Localization of herpessimplex
virus type 1 nucleocapsid polypeptides byimmune elec-tronmicroscopy. J. Gen. Virol. 54:39-46.
35. Wagner, E. 1983. Transcription patterns in HSV infec-tions, p. 239-270. In G. Klein (ed.), Advances in viral oncology, Vol. 3. Raven Press, New York.
36. Wagner, E., K. Anderson,R.Costa, G.Devi,B.Gaylord, L.Holland, J. Stringer, andL. Tribble. 1981. Isolationand characterization of HSV-1mRNA, p. 45-67. InY. Becker (ed.), Developments in molecular virology, vol. 1: Her-pesvirus DNA. MartinusNijhoff,B.V., The Hague.
on November 10, 2019 by guest
http://jvi.asm.org/
ERRATUM
High-Resolution Characterization of
Herpes
Simplex Virus
Type 1 Transcripts
Encoding Alkaline Exonuclease
and
a
50,000-Dalton
Protein
Tentatively
Identified
as a
Capsid
Protein
R. H.COSTA, K. G.DRAPER, L. BANKS, K. L. POWELL. G.COHEN, R. EISENBERG, ANDE. K. WAGNER
Department of MolecularBiologyand Biochemistry, Universit'ofCalifornia, Irvine. Californiia92717;DepacrtmentofMicrobiology (Virology), Universityof Leeds, LeedsLS2 9JT, England:andDepairtnentofMicrobiologyatndCenterforOrial HealthResearch, Dental
Medicine, and Departmentof Pathobiologv. SchoolofVeterinaryMedicine, Universityof Pennsylvania, Phildelpliti, Pensylvani 19104
Volume 48, no. 3, p. 591-603: In this paper the authors incorrectly quoted Lemaster and Roizman (S. Lemaster and B. Roizman, J. Virol. 35:798-811, 1980 [20]) as locating VP-19C in the region in question. VP-19Chas been located elsewhere on the HSV-1 genome (D. K. Braun, W. Batterson,and B. Roizman, J. Virol., in press). The protein located by Lemaster and Roizman is VP-18.8.