Copyright © 1998, American Society for Microbiology. All Rights Reserved.
An Enhanced Packaging System for Helper-Dependent
Herpes Simplex Virus Vectors
TOM A. STAVROPOULOSANDCRAIG A. STRATHDEE*
Gene Therapy and Molecular Virology Group, The John P. Robarts Research Institute, London, Ontario, Canada N6A 5K8, and Department of Microbiology and Immunology,
University of Western Ontario, London, Ontario, Canada N6A 5C1
Received 27 March 1998/Accepted 9 June 1998
Helper-dependent herpes simplex virus (HSV) vectors (amplicons) show considerable promise to provide for long-term transduced-gene expression in most cell types. The current packaging system of choice for these vectors involves cotransfection with a set of five overlapping cosmids that encode the full HSV type 1 (HSV-1) helper virus genome from which the packaging (pac) elements have been deleted. Although both the helper virus and the HSV amplicon can replicate, only the latter is packaged into infectious viral particles. Since the titers obtained are too low for practical application, an enhanced second-generation packaging system was de-veloped by modifying both the helper virus and the HSV amplicon vector. The helper virus was reverse engi-neered by using the original five cosmids to generate a single HSV-bacterial artificial chromosome (BAC) clone in Escherichia coli from which the pac elements were deleted to generate a replication-proficient but packaging-defective HSV-1 genome. The HSV amplicon was modified to contain the simian virus 40 origin of replication, which acts as an HSV-independent replicon to provide for the replicative expansion of the vector. The HSV amplicon is packaged into infectious particles by cotransfection with the HSV-BAC helper virus into the 293T cell line, and the resulting cell lysate is free of detectable helper virus contamination. The combination of both modifications to the original packaging system affords an eightfold increase in the packaged-vector yield.
Herpes simplex virus (HSV) is a neurotropic herpesvirus that can establish lifelong infections in its human host through latent maintenance in the ganglia of sensory neurons. The virus is relatively well characterized, with a genome of;155 kb that is maintained as a concatemerized circular or linear episome in infected cells (27). Because of their wide host range, efficient infection, long-term persistence, capacity to accommodate large amounts of foreign DNA, and ability to deliver genes to post-mitotic cell types, considerable effort has been expended to develop gene transfer vectors that can exploit the natural bi-ology of HSV (1, 10, 19). Both dependent and helper-independent vectors are currently in wide use. Helper-indepen-dent HSV vectors contain the complete viral genome but have deletions in one or more essential viral genes and can be used to express three or more cDNAs by replacing other, nonessen-tial viral genes (17). Helper-independent HSV vectors thus have a replication-defective viral phenotype and can establish a productive infection only on complementing cell lines that express the corresponding deleted viral gene (5). The major problem associated with this class of vectors, in general, is that there is leaky or inappropriate expression of some of the im-mediate-early and early HSV genes in noncomplementing cell types, which ultimately results in death of the infected cell even though no viral replication has occurred (14–16, 42). Such cytotoxicity can be completely eliminated when all of the im-mediate-early HSV genes are deleted, but this results in very low levels of transduced reporter gene expression (28, 29). Thus, entry into the latent cycle may be mandatory for gene expression and maintenance of helper-independent HSV vec-tors, effectively limiting their use to neuronal cell types. Even so, the factors controlling the complete latency-associated
shutdown of viral gene expression have not been clearly de-fined for HSV, and limited progress has been made in bypass-ing this shutdown to facilitate long-term transduced-gene ex-pression (18, 35, 36).
Helper-dependent HSV vectors (commonly known as HSV amplicons) contain only the cis elements required for HSV replication and packaging, the oriSand pac elements,
respec-tively (8, 34) (Fig. 1B). Because the size of the vector backbone is only a small fraction of that of the HSV genome, usually
,10%, HSV amplicons have the potential to express a large number of genes or cDNAs. Moreover, since they contain no virus-encoded genes, these vectors have the potential to pro-vide long-term gene expression in transduced cells by obviating the latency-associated shutdown of HSV gene expression and are thus well positioned to take full advantage of the ability of HSV to infect virtually every type of cell (2, 8, 11). The first practical packaging system for HSV amplicons utilized a rep-lication-defective with HSV ICP4 deleted as a helper virus to provide the necessary viral replication and packaging functions (9). In this system, the amplicon vector is transfected into an
ICP4-complementing cell line, which is then superinfected with
thehelpervirus,andtheresultinginfectiousparticlesareharvest-ed following growth of the virus. Although this approach can yield recombinant-virus titers approaching 109PFU/ml, unless
there is a mechanism in place to provide for the selective replication and packaging of the amplicon vector over that of the helper virus, the transducing lysates produced are heavily contaminated with the helper virus (9, 20). Even when selective replication and packaging of the amplicon vector is provided for, the helper virus represents 10 to 25% of the total virus yield produced (25, 44). Similar to the situation with helper-independent vectors, this helper virus contamination leads to significant delivery-associated cytotoxicity in transduced cells. Although this problem can potentially be eliminated through the use of less cytotoxic forms of HSV to package amplicon * Corresponding author. Mailing address: Gene Therapy and
Mo-lecular Virology Group, The John P. Robarts Research Institute, P.O. Box 5015, 100 Perth Dr., London, Ontario, Canada N6A 5K8. Phone: (519) 663-5777, ext. 4032. Fax: (519) 663-3789. E-mail: [email protected].
7137
on November 9, 2019 by guest
http://jvi.asm.org/
vectors, such mutants typically grow much more slowly and the packaged-amplicon yield is correspondingly lower (38).
A second-generation packaging system was recently devel-oped for helper-dependent HSV vectors that yields transduc-ing lysates free of helper virus contamination, maktransduc-ing it the packaging system of choice for HSV amplicons (7). The system is centered around a set of five overlapping HSV-1 cosmid clones that together encode the complete wild-type viral ge-nome (4) (Fig. 1A). By specifically deleting the pac element in the repetitive a sequences of two of the cosmids, the cosmid set is rendered packaging defective, and when it is cotransfected into mammalian cells along with an HSV amplicon vector, only the latter is packaged into mature viral particles. The deletion of the pac elements addresses the requirement for selective packaging of the vector over the helper virus. However, it does not provide for any selective replication of the amplicon vector, and this may be why the overall yield of packaged vector particles is quite low, usually,106/ml.
In this report, we describe the development of an enhanced second-generation packaging system for helper-dependent HSV vectors, which we achieved by modifying both the pack-aging-defective helper virus and HSV amplicon vector compo-nents of the current system. We modified the helper virus by cloning the entire packaging-defective HSV-1 genome as a single infectious plasmid in bacteria. We modified the ampli-con vector by incorporating the simian virus 40 (SV40) origin of replication, which acts as an HSV-independent replicon to provide for the replicative expansion of the vector prior to packaging into infectious HSV-1 particles. The combination of both modifications affords an eightfold increase in the pack-aged-vector yield and provides sufficient material in benchtop-scale production to enable in vivo studies for most applica-tions.
MATERIALS AND METHODS
Cell lines and plasmids.The BHK(TK2) cell line (hamster kidney) was
pro-vided by Paul Johnson and Theodore Friedman (15). The SV40
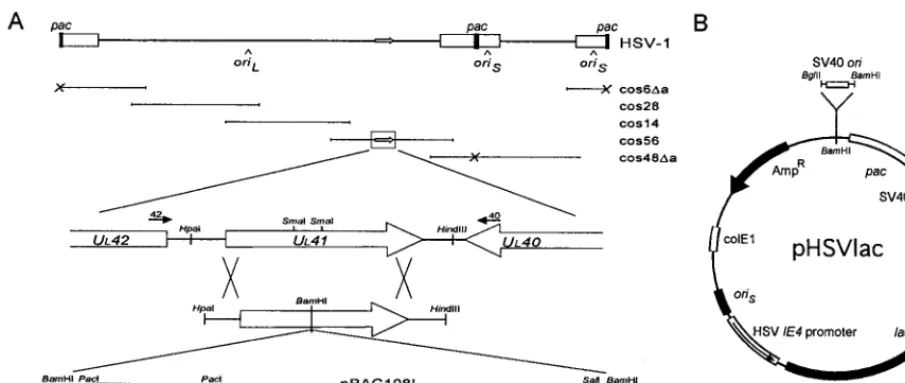
T-antigen-positive 293T-17 cell line (human embryonic kidney) was provided by Doug Bell and David Baltimore and was cultured with 400mg of Geneticin/ml (24). The Vero cell line (African green monkey kidney) was provided by Dora Ho. Each cell line was cultured in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum and was split 1:4 every 3 to 4 days by standard methods. The PC12 cell line (rat adrenal pheochromocytoma) was cultured in Dulbecco mod-ified Eagle medium supplemented with 5% fetal calf serum and 5% horse serum and was differentiated by adding murine nerve growth factor to a final concen-tration of 50 ng/ml (Harlan Bioproducts for Science Inc., Madison, Wis.) (39). The pUL41 plasmid, encoding the HSV-1 vhs gene, was provided by James Smiley (31). The pa4bgal/ori vector, which contains the HSV-1 pac element, was provided by Dora Ho (12). Bacterial artificial chromosome (BAC) cloning vector pBAC108L and BAC host strain Escherichia coli HS996 were provided by Melvin Simon and Hiroaki Shizuya (30). HSV-1 cosmid set C was provided by Andrew Davison, and the modified cos6Da and cos48Da constructs and the pHSVlac vector were provided by Cornel Fraefel and Alfred Geller (4, 7). The latter was modified to include the minimal SV40 ori element by ligating the BglII-HindIII fragment of the pGL3-Promoter vector (Promega Inc., Madison, Wis.) into the unique BamHI site to generate pHSVlacOri (Fig. 1B).
Construction of the targeting vector.The BAC vector was constructed by subcloning a cassette containing the HSV-1 pac element and theafragment of the bacterial lacZ gene into the SalI site of pBAC108L as a SalI-XhoI fragment (Fig. 1A). The HSV pac cassette was constructed by using the pcDNAII expres-sion vector (Invitrogen Inc., La Jolla, Calif.), in which the two NsiI sites within the multiple cloning site were fused, the NaeI site was converted to SalI, and the TfiI site was converted to PacI-XhoI by using oligonucleotide linkers. The HSV-1 pac element from pa4bgal/ori was ligated into the SalI site as a SalI-XhoI fragment by using oligonucleotide linkers, after which the SalI site was converted to SalI-BamHI-PacI. The completed BAC construct was subcloned between the two SmaI sites of pUL41 as a linearized BamHI fragment. To accommodate the BAC, an oligonucleotide linker was used to first convert the SmaI sites of pUL41 to BamHI sites, such that the BAC can be excised as an intact fragment. The targeting vector was linearized by digestion with HindIII prior to cotransfection into BHK cells with the HSVDa cosmid set at a ratio of 1:5.
Packaging of HSV amplicons.All plasmid, cosmid, and BAC DNA was pre-pared by using a modified alkaline lysis protocol, followed by purification over a proprietary ion-exchange column in accordance with the manufacturer’s (Qiagen Inc., Valencia, Calif.) protocols. The pBAC-HSV constructs were further treated to remove contaminating bacterial endotoxin by using a proprietary reagent (Qiagen Inc.) and stored in small aliquots at220°C to minimize shearing of the DNA due to repeated freeze-thawing. The HSV-1 cosmids were digested with PacI and repurified by phenol-chloroform extraction prior to transfection. The pHSVlac vectors were packaged into infectious particles by cotransfection with either the HSVDa cosmid set or pBAC-V2 at a ratio of 1:4 by using a proprietary cationic liposome formulation (LipofectAMINE) in accordance with the manu-facturer’s (Life Technologies Inc., Bethesda, Md.) protocols. Cells were
trans-FIG. 1. Recombinant HSV-1 constructs. (A) Structure of the UL41-BAC targeting vector. At the top is a schematic representation of the HSV-1 genome and the correspondingDa cosmid set, comprised of cos6Da, cos48Da, cos14, cos28, and cos56. The UL41 gene is located within cos56, as shown enlarged in the middle. Open reading frames are indicated by open arrows. The bottom shows the structure and orientation of the UL41-BAC targeting construct, including the pac cassette containing the HSV-1 pac element and theafragment of the bacterial lacZ gene. The large crosses indicate the regions of homology between the targeting vector and cos56 that will facilitate homologous recombination to generate a packaging-proficient recombinant virus. The relative location and orientation of the oligonucleotide primers used for PCR are as indicated. Primers 40 and 42 are located outside of the region of homology with the UL41-BAC targeting construct. (B) Structure of the pHSVlac amplicon vector. The HSV-1 oriSand pac elements provide for helper-dependent packaging of the vector. Expression of the bacterial lacZ reporter gene is controlled by the HSV-1 IE4 promoter and SV40 polyadenylation signals, as indicated. The SV40 ori element was cloned into the unique BamHI site adjacent to the HSV pac element.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.75.528.71.262.2]fected with 2mg of DNA plus 15ml of LipofectAMINE in one well of a six-well culture dish when they reached 80 to 90% confluence. Following transfection, N,N9-hexamethylene-bis-acetamide (Sigma Chemical Co., St. Louis, Mo.) was added to a final concentration of 2 mM to stimulate immediate-early viral gene expression (23). The medium was changed and fresh N,N9 -hexamethylene-bis-acetamide was added 24 h following transfection, and the cultures were grown until evidence of the viral cytopathic effect was noted (usually a further 1 to 3 days). Viral particles were harvested by scraping the cells and supernatant into a sterile tube and lysing the cells in two rapid freeze-thaw cycles. Cellular debris was removed by centrifugation at 1,0003g for 10 min, and the viral particles in the supernatant were aliquoted for storage at280°C (11). The infectious-vector and/or infectious-virus yield was determined by assaying serial dilutions for b-galactosidase activity by using 5-bromo-4-chloro-3-indolyl-b-D
-galactopyrano-side (X-Gal) histochemistry (21) or for plaque-forming activity (11), respectively, on Vero cells.
Analysis of viral DNA.Infected cells from eight P150 culture dishes were harvested by scraping and centrifugation at 1,000 3g for 10 min and then resuspended in 8 ml of TE buffer (10 mM Tris-HCl [pH 7.8], 100 mM EDTA) and lysed after adding a further 8 ml of TE buffer supplemented with 2% Triton X-100 by using a Dounce homogenizer. Cellular debris was removed by centrif-ugation at 2,0003g for 10 min, and capsids were pelleted by centrifugation at 17,0003g for 90 min. The capsids were resuspended in 0.5 ml of TEN buffer (10 mM Tris-HCl [pH 7.8], 10 mM EDTA, 150 mM NaCl), and the viral capsids were removed by digestion with 2-mg/ml proteinase K and 0.1% (final concentration) sodium dodecyl sulfate at 60°C for 1 h, followed by phenol extraction and dialysis against three changes of TE buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA). For ligation reactions, 100 ng of DNA was utilized in a 20-ml reaction volume to promote recircularization. After incubation overnight at 14°C, 5ml of the reac-tion mixture was electroporated into competent HS996 bacterial cells by using standard protocols. For field inversion gel electrophoresis, 100 ng of DNA was loaded onto a 1.0% agarose gel in 0.53Tris-borate-EDTA (TBE), and the gel was run for 20 h at 200 V and 14°C. Forward pulse times were ramped from 0.9 to 6.3 s with a forward-to-reverse pulse time ratio of 3:1.
PCR amplifications.HSV-specific primers were targeted to the UL40 gene
(primer 40 [ACCATAGCCAATCCATGACC]) and the UL42 gene (primer 42
[GTCGTGAGGAAGAACTTGAGG]) and were designed to amplify across the entire UL41 gene. BAC-specific primers were targeted to the forward region
(primer BF [TATTGACATGTCGTCGTAACC]) and the reverse region (prim-er BR [ATGTCGGCAGAATGCTTAATG]) flanking the PacI cassette and, in conjunction with primers 40 and 42, respectively, were designed to amplify across the sites of BAC integration into the HSV-1 UL41 gene. A high-processivity Taq
polymerase cocktail (Expand Long Template PCR System; Boehringer Mann-heim GmbH, MannMann-heim, Germany) was used for all reactions. The annealing temperature for all primer pairs was 60°C, with 100 ng of BAC template DNA or 30 ng of cosmid template DNA in each reaction mixture. Dimethyl sulfoxide was included to a final concentration of 10% for those reactions that involved exten-sion across the GC-rich HSV-1 pac element, but otherwise, the thermal profile and all other parameters for amplification were in accordance with the manu-facturer’s recommendations. PCR products were analyzed by agarose gel elec-trophoresis by standard methods.
RESULTS
Cloning strategy. The development of a packaging system for helper-dependent HSV vectors that could provide lysates that are free of contaminating helper virus was a significant breakthrough in terms of facilitating the efficient, nontoxic delivery of infectious vector particles (7). However, there are a number of limitations inherent to this system that make it technically demanding to use and that limit the ultimate pack-aged amplicon vector yield. (i) Packaging requires the trans-fection of five overlapping HSV-1 cosmid clones, which must recombine to form a functional HSV-1 genome prior to repli-cation. (ii) Some of the cosmid clones are unstable when prop-agated in bacteria, and the preparation of high-quality DNA for transfections is therefore somewhat laborious. (iii) Trans-fection efficiencies approaching 100% are required to maxi-mize the packaged amplicon vector yield. We hypothesized that it should be possible to surmount these limitations by cloning the entire packaging-defective HSV-1 genome as a single plasmid DNA by using a BAC cloning vector (30). This would provide a technically simple and much more efficient method of recon-stituting the helper virus in the packaging cell. The BAC vector was developed in response to the need to stably propagate large genomic DNA fragments in bacteria, and thus, it is likely
that the cloned HSV-1 genome will be more stable as a BAC than in the presently used cosmids.
The approach we pursued was to reverse engineer a single HSV-BAC clone from the five original HSV-1 cosmid clones with the pac elements already deleted and is based on the ability of the HSV-1 cosmid set to generate a functional viral genome through recombination in mammalian cells (4). Since it has been established that only one pac element is required for packaging of the HSV-1 genome (7) and that pac elements retain their function when placed at an alternative locus (3, 32), we inferred that it should be possible to reconstitute a packaging-proficient HSV-1 genome from theDa cosmid set by
providing a suitable pac element. Our strategy was to generate a functional virus from the HSVDa cosmid set by targeting a
BAC vector containing a single pac element to a nonessential HSV-1 gene through homologous recombination. The BAC vector facilitates the subsequent cloning of the resulting re-combinant viral genome in bacteria as a single construct. The
pac element can then be removed from this construct in vitro
to generate a packaging-defective HSV-1 clone that can be used as a helper virus for the packaging of HSV amplicon vectors.
Generation of an infectious HSV-1 clone in a BAC vector.
We selected the HSV-1 UL41 gene as the site of integration of
the BAC vector. The UL41 gene product is a tegument protein
that is incorporated into mature viral particles and is involved in the degradation of host cell mRNA to provide the viral host shutoff (vhs) function (31). Since the ultimate use of the helper virus will be to provide efficient delivery of the packaged HSV amplicon, we expect that the elimination of the potentially toxic vhs function from the infectious particle should have a positive effect on survival of the transduced cells. We designed a UL41 targeting vector that contained an HSV pac cassette
within a standard BAC cloning vector and then transfected it into BHK cells along with the HSVDa cosmid set (Fig. 1A).
The resulting recombinant HSV-BAC viral progeny were ex-panded on Vero cells to obtain sufficient quantities for purifi-cation of the viral genomic DNA. We elected not to clone out individual viral recombinants at this stage but rather to select for the fittest recombinants in the population by growing them as a single pool. This strategy ensures that the fittest viral prog-eny during growth in vitro will thus be overrepresented when cloned out in bacteria. Since the recombinant HSV-BAC viral progeny have the BAC cloning vector integrated into the UL41
gene, these were cloned simply by recircularizing the linear viral genome and electroporating the DNA into BAC host strain HS996.
We recovered three independent clones and characterized them by restriction endonuclease fingerprinting with BamHI to determine which contained a complete and intact HSV-1 genome. Since the three had very similar patterns (data not shown), we first determined if any were still infectious and able to generate plaques when transfected back into BHK cells. One of the clones was able to produce a functional virus in this assay and was designated pBAC-V1. We assessed the relative efficiency of plaque formation by the pBAC-V1 clone by com-paring it to that of the original, packaging-proficient HSV-1 cosmid set. The DNA constructs were transfected into BHK cells in a single well of a six-well culture dish, and after 4 days, the viral particles were harvested and the total virus yield was determined by assaying serial dilutions for plaque formation on Vero cell monolayers. Under these conditions, the pBAC-V1 and HSV cosmid set yielded 5.03103and 6.03102PFU/
ml, respectively. Thus, despite the fact that it has a deletion at the UL41 locus and contains only a single pac element, the
pBAC-V1 clone produced eightfold more virus particles when transfected into BHK cells than did the HSV cosmid set, which
on November 9, 2019 by guest
http://jvi.asm.org/
contains an intact viral genome that includes two pac elements. This result substantiates our initial expectation that an intact viral genomic fragment should be intrinsically more efficient at generating a functional virus than five overlapping genomic fragments.
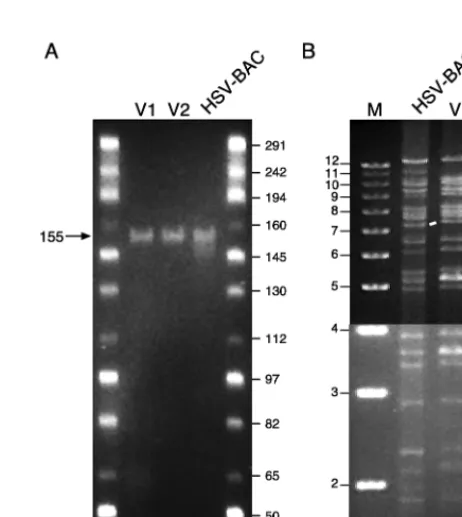
We used three independent analyses to determine if the pBAC-V1 clone contains the full HSV-1 genome. First, we characterized the site of integration of the BAC vector into the HSV-1 genome by using PCR amplification (Fig. 2). Amplifi-cation across the entire UL41 locus by using the 42-40 primer
set yielded a 4.3-kb product in cos56 which corresponds to the intact UL41 gene and an 11.2-kb product in pBAC-V1 which
corresponds to integration of the complete BAC targeting con-struct within the UL41 gene. Furthermore, amplification across
the integration junctions of the BAC targeting construct using the 40-BF and 42-BR primer sets yielded products of 2.2 and 3.0 kb, respectively, confirming that the targeting construct correctly localized the BAC in the intended location within the
UL41 gene. Second, we analyzed the size of the PacI-digested
pBAC-V1 clone by using field inversion gel electrophoresis and found it to be identical to that of the genomic DNA of the original pooled HSV-BAC recombinant virus at;155 kb (Fig. 3A). Third, we compared the BamHI restriction endonuclease fingerprint of the clone to that of the genomic DNA of the original pooled HSV-BAC recombinant virus (Fig. 3B). Al-though a few minor differences in the patterns were noted, these are likely attributable to heterogeneity in the pooled virus sample. For example, note that the fragments at 11.0, 10.5, 9.0, and 8.5 kb in the pooled HSV-BAC viral DNA are present at half of the expected intensity, whereas in the pBAC-V1 sample, only the 10.5- and 9.0-kb fragments are present but, in this case, at full intensity. These differences are due to recombination in the repeat regions of the HSV-1 ge-nome that generate four alternative isomers of the viral DNA sample (3, 26). The BamHI fingerprinting analysis confirms that there is equal representation of all four isomers in the pooled viral DNA sample and that only one of the isomers is represented in the pBAC-V1 clone. The results of all three analyses are consistent with the conclusion that the pBAC-V1 clone contains an otherwise intact HSV-1 genome in which the
UL41 gene has been inactivated. We have also done similar
analyses by using pBAC-V1 clones that have been serially
sub-cultured in bacteria and have never recovered any rearrange-ments (data not shown), suggesting that the HSV-1 genome is completely stabilized in the BAC vector. Thus, we proceeded to the next step in developing a helper virus-free packaging system by rendering pBAC-V1 packaging defective through deletion of the single pac element present.
[image:4.612.69.271.70.223.2]Generation of a packaging-defective HSV-1 clone.The BAC cloning strategy we developed was designed to facilitate the efficient removal of the single pac element in the pBAC-V1 clone. The HSV pac cassette is flanked by PacI restriction endonuclease sites, and since PacI does not cut anywhere else in the BAC vector or in the HSV-1 genome, the cassette can be excised by digestion with PacI, followed by recircularization of the construct. To facilitate the identification of clones with pac deletions, we incorporated theafragment of the bacterial lacZ gene into the original PacI cassette. The pBAC-V1 clones that
FIG. 2. Integration of the BAC vector into the HSV-1 UL41 gene. PCR
products were generated from HSV cos56 and pBAC-V1 and pBAC-V2 with the indicated primer pairs and separated through a 0.8% agarose gel. The loss of 1.0 kb from the PCR products derived from pBAC-V2 is due to excision of the pac cassette from this clone. The marker used in lane M is the 1-kb Plus Ladder (Life Technologies Inc.). Molecular sizes are shown on the left in kilobases.
FIG. 3. Analysis of recombinant BAC clones. (A) Analysis of recombinant and cloned HSV-1 constructs by field inversion gel electrophoresis. Purified viral genomic DNA from the pooled HSV-BAC recombinants was analyzed without further processing, whereas the pBAC-V1 and pBAC-V2 clones were linearized by digestion with PacI prior to loading onto the gel. The arrow indicates the position of the linearized HSV-1 genome at;155 kb. The 1-kb PacI fragment corresponding to the pac cassette excised from pBAC-V1 is not resolved in this analysis. The marker used in lane M is Midrange PFG Marker I (New England BioLabs, Beverly, Mass.), and the corresponding sizes in kilobases are indicated on the right. (B) Restriction endonuclease fingerprinting of recombinant and cloned HSV-1 constructs. DNA from the various constructs was digested with BamHI and resolved by electrophoresis through a 0.6% agarose gel. In order to provide adequate contrast for the different fragment sizes down the entire gel, the image shown is a composite of two different photographic exposures. The closed arrows indicate the 11.0-, 10.5-, 9.0-, and 8.5-kb fragments corresponding to the different isomers in the pooled recombinant HSV-BAC genomes, and the larger closed arrows indicate the 10.5- and 9.0-kb fragments corresponding to the isomer cloned in the pBAC-V1 and -V2 constructs. The open arrow indicates the 7.5-kb fragment corresponding to the excised BAC vector in the pBAC-V1 sample, and the white lines indicate changes in the size of this fragment in the pooled recombinant HSV-BAC and pBAC-V2 samples. In pBAC-V2, the BAC vector is reduced in size by 1 kb due to removal of the pac cassette, and in the HSV-BAC sample, the BAC vector is reduced in size by 0.2 kb due to cleavage of the recombinant viral genome within the pac element. The marker used in lane M is the 1-kb Plus Ladder (Life Technologies Inc.), and the sizes of the corresponding bands are indicated in kilobases on the left.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.310.541.256.514.2]contain the cassette are blue on X-Gal-containing plates, whereas clones that have lost the cassette are white. We re-covered a number of such clones by using this simple screen and characterized two of them with respect to their BamHI restriction endonuclease fingerprints to determine if the HSV-1 genome was intact. Except for a 1-kb reduction of the 7.5-kb BAC-derived fragment corresponding to the loss of the HSV pac cassette, both clones were identical to the parental pBAC-V1 construct. The BamHI fingerprint of one of these, designated pBAC-V2, is shown in Fig. 3B. Analysis of the PacI-linearized clone by field inversion gel electrophoresis confirms that its size is the same as that of pBAC-V1 (Fig. 3A).
We further characterized pBAC-V2 by using the same meth-odology described for the parental pBAC-V1 clone. PCR am-plification across the entire UL41 locus by using the 42-40
prim-er set yielded a 10.2-kb product, and amplification across the integration junctions of the BAC targeting construct by using the 42-BR primer set yielded a 2.0-kb product for pBAC-V2 (Fig. 2). The loss of 1.0 kb from the PCR products derived from pBAC-V2 in comparison to pBAC-V1 is consistent with the loss of the HSV pac cassette from this clone. In addition, we transfected pBAC-V2 into BHK cells to determine if it was still infectious and able to generate plaques, as described pre-viously for pBAC-V1 and the HSV cosmid set. As expected from the combined data indicating that the pac element was deleted from this clone, no infectious HSV-1 particles were produced. These results are consistent with the conclusion that pBAC-V2 contains a replication-competent but packaging-de-fective HSV-1 genome and that this construct can be used to package amplicon vectors that will be free of helper virus con-tamination.
Packaging of an HSV amplicon vector into infectious parti-cles.Since the packaging-defective HSVDa cosmid set can be
used as a helper virus to package HSV amplicons (7) and the re-sulting cellular lysates remain free of helper virus contamina-tion, we evaluated the ability of the pBAC-V2 clone to function in a similar role. When the prototypical pHSVlac amplicon vector was used as a substrate, pBAC-V2 was able to provide appropriate helper functions and package the vector to a titer of 8.03104blue-forming units (BFU)/ml and with no
detect-able helper virus contamination. In comparison with the HSV
Da cosmid set, which yielded a titer of 2.03104BFU/ml in a
parallel transfection, pBAC-V2 was fourfold more proficient at packaging the pHSVlac vector into infectious particles. We have repeated this experiment many times with a variety of cell lines and with a number of different HSV amplicon vectors, and although there is considerable variation in the titer be-tween experiments, pBAC-V2 consistently yields two- to eight-fold more packaged vector than does the HSVDa cosmid set.
BHK cells were used in these experiments because they rou-tinely provide transfection efficiencies surpassing 90% in our studies. However, as described for the cosmid packaging set, any cell line can be used for amplicon packaging (7). We have found that similar high transfection efficiencies can be achieved by using 293T cells (24), whereas those in other laboratories utilize the Vero-based 2-2 cell line (7, 33). Regardless of the cell line used, we have never observed any difference in trans-fection efficiency for either pBAC-V2 or the HSVDa cosmid
set when it is used as a helper virus (data not shown). It is unlikely that the increased amplicon packaging efficiency of the former can be attributed to differences in transfectability, al-though given the greater technical difficulties in preparing DNA from the HSVDa cosmid set, this remains a possibility.
A more likely interpretation of this finding is that the packaged amplicon vector yield of either system is directly related to the infectivity of the transfected helper virus DNA. The pBAC-V2
construct is therefore more efficient than the HSVDa cosmid
set because it does not require any recombination events to generate the replication-competent viral genome that is re-quired to package amplicon vectors.
We utilized PC12 cells to examine the ability of infectious particles generated by the pBAC-V2 helper virus genome to deliver the packaged pHSVlac vector to postmitotic cells. When grown in media containing nerve growth factor, PC12 cells exit the cell cycle and differentiate to resemble sympathetic neu-rons in many aspects of cellular physiology (39). We compared the plating efficiencies of the packaged pHSVlac vector on differentiated PC12 cells and Vero cells. Since theb -galacto-sidase transducing titers were identical, we concluded that the pBAC-V2 helper virus genome provides all of the necessary HSV-1 proteins for efficient infection of neuronal cell types. Moreover, the vector-transduced cells show no detectable signs of toxicity at 3 days postinfection (Fig. 4). In this experiment, the lowest dilution of the packaged pHSVlac vector that was plated resulted in a multiplicity of infection of 0.8 particle/cell, such that the majority of transduced PC12 cells were targeted by a single infectious particle. Because the HSV-1 vhs function is deleted from the pBAC-V2 helper virus genome, we expect that the infectious HSV-1 particles generated will be tolerated equally well at much higher multiplicities of infection, for ex-ample, as can occur in vivo during direct injection of packaged vector material into the mammalian central nervous system. We now have data from rat and hamster models using a variety of different amplicon vectors that support this premise (unpub-lished data).
An alternative viral replicon enhances HSV amplicon vector packaging.The inefficiency of first-generation HSV packaging systems is due to their inability to provide for the selective replication and packaging of the amplicon vector. The HSV cosmid set and HSV-BAC packaging systems work in part because they specifically address the latter deficiency through the use of a packaging-defective helper virus. Since they do not select for replication of the amplicon vector, we postulated that the packaged amplicon vector yield may be limited by ineffec-tive competition with the helper virus for HSV-specific repli-cation factors. If this is the case, then a simple solution is to specifically increase the copy number of the amplicon vector relative to that of the helper virus, in essence ensuring that subsequent replication is skewed to favor the vector rather than the helper virus. Our approach was to use an alternative, HSV-independent replicon to afford such a competitive advan-tage to the amplicon and thus increase the overall yield of
FIG. 4. Transduced reporter gene expression in postmitotic cells. Differen-tiated PC12 cells were infected at a multiplicity of;0.8 in a single well of a P24 culture dish, and 2 days later, the cells were fixed and stained for pHSVlac-derivedb-galactosidase reporter gene activity by using X-Gal histochemistry. The black and white arrows indicate positive and negative staining of cells, respectively. The boxed area in panel B indicates the location of the field shown at a higher magnification in panel A.
on November 9, 2019 by guest
http://jvi.asm.org/
packaged vector particles. It is well established that the SV40 origin of replication can facilitate the selective amplification of virtually any plasmid DNA in cells that express the correspond-ing SV40 T antigen (40). Accordcorrespond-ingly, we utilized the SV40 system to determine if this would have a similar effect on amplicon packaging.
The pHSVlac vector was modified to include the minimal elements of the well-characterized SV40 ori element, which includes the basal SV40 early promoter but not the enhancer (Fig. 1B). We compared the efficiency of packaging of pHSV-lac and that of modified pHSVpHSV-lacOri in BHK and 293T cells (Table 1). In the T-antigen-positive 293T cell line (24), the packaged amplicon vector yield is increased almost twofold when the vector contains the SV40 ori element. This increase is not seen in the T-antigen-negative BHK cell line, where the pHSVlac vector is packaged more efficiently than modified pHSVlacOri. In independent transient transfection experi-ments, we determined that the copy number of pHSVlacOri is increased fourfold over that of pHSVlac by 48 h posttransfec-tion in 293T cells but is unchanged in BHK cells (data not shown). Therefore, we concluded that the enhanced packaging of pHSVlacOri in 293T cells is due to T-antigen-dependent prereplication of the vector and that this relatively small in-crease in vector copy number effectively overcomes the slight packaging disadvantage of pHSVlacOri observed in BHK cells. Since the vector copy number can be increased in other ex-pression systems by several orders of magnitude by using the SV40 ori element (40), this suggests that further increases in packaging efficiency may be attainable by increasing the repli-cative efficiency of the added replicon, for example, by increas-ing the activity of the SV40 ori element by incorporatincreas-ing the full SV40 enhancer or by incorporating a more efficient viral replicon into the vector.
DISCUSSION
The development of gene-based therapies is currently being pursued in fields as diverse as infectious disease, autoimmune disease, cardiovascular disease, and neurodegenerative dis-ease. The availability of an efficient gene delivery and expres-sion system is an essential enabling technology that unites all of these applications, and as a result, this has been an area of intense research in recent years. HSV-based vectors show con-siderable promise in this regard because of their wide host rang, efficient infection, long-term persistence, capacity to ac-commodate large amounts of foreign DNA, and ability to deliver genes to postmitotic cell types (5, 10, 13). After further optimization of culture and transfection conditions and only a
modest upscaling of our methods, we have succeeded in re-producibly obtaining titers of 107 transducing particles/ml of
lysate for a variety of different HSV amplicon vectors by using the pBAC-V2 helper construct. Thus, the HSV packaging sys-tem is now efficient and economical enough to provide suffi-cient material for most in vivo studies, and we anticipate that it will be possible to extend these to include expression of a therapeutic gene in various animal models of disease. In this regard, future developments will likely lie in refinements to the vector backbone itself in order to stabilize the HSV amplicon and thus afford extended transduced-gene expression. One ap-proach that we and others have taken is to incorporate an alternative viral replicon, for example, the episomal replicon of the Epstein-Barr virus (37, 41) or the integrating replicon from the adeno-associated virus (6), and it is likely that additional hybrid HSV amplicon vectors are already in development in other laboratories. Given the recent improvements in helper-independent HSV vectors that eliminate all delivery-associated cytotoxicity (28, 29), it will be interesting to compare the rel-ative efficiency of long-term transduced-gene expression from this platform with that obtained by using the newer hybrid amplicon vector systems.
The ability of the SV40 ori element to increase the pack-aged-vector yield in our packaging system stands in contrast to data obtained by using a fully functional HSV helper virus, where the incorporation of the SV40 ori element has no effect on amplicon packaging (43). Thus, the ability of prereplication to increase the packaged-vector yield may be context specific, for example, dependent upon the use of a packaging-deficient helper virus and/or a specific HSV amplicon. For example, the prereplication by the SV40 ori element may be effective only under those conditions in which replication of the HSV helper virus (and thus the HSV amplicon) is suboptimal, as is the case when a replication-defective or packaging-deficient HSV help-er virus is used. In support of this view, we have incorporated the minimal SV40 ori element into other HSV amplicon vec-tors that our laboratory has engineered and have found that it is more effective in those vectors that are packaged relatively inefficiently in comparison to pHSVlac and can increase pack-aged-vector yields by as much as 10-fold in such cases (unpub-lished data).
The strategy we developed for cloning of the HSV-1 genome as a single infectious BAC should be applicable to other DNA viruses. Indeed, during the course of our experiments, a re-markably similar approach was described for the murine cyto-megalovirus (mCMV), a herpesvirus related to HSV (22). In this case, the experimental goal was to develop a system in which the mCMV genome could be manipulated in bacteria such that the phenotype of viral mutants could be defined without the requirement for growth in a mammalian cell that is a prerequisite in conventional mutagenesis protocols. In such a strategy, it is imperative that the cloned viral genome be stable in bacteria, such that the incidence of unwanted or cryptic mutations in other viral genes is minimized, since these would confound interpretation of the resulting viral phenotype. In this regard, the growth kinetics of the HSV-BAC virus and the virus derived from the infectious pBAC-V1 clone are identical. Moreover, we have never detected any rearrangements in the pBAC-V2 clone following serial passage in bacteria, suggesting that the cloned HSV genome appears to satisfy this criterion. Thus, we expect that a mutagenesis scheme similar to that described for the cloned mCMV genome can be applied to HSV-1. The fact that we were able to generate a packaging-deficient but otherwise completely functional viral genome il-lustrates the power of such an approach, since it is otherwise impossible to propagate such a recombinant virus in mamma-TABLE 1. Enhanced packaging of an HSV amplicon vector
Vector Cell linea Vector titer b
(104BFU/ml
of lysate) Virus titer
c
pHSVlac 293T 1.5 ,10
pHSVlacOri 293T 2.7 ,10
pHSVlac BHK 2.4 ,10
pHSVlacOri BHK 1.8 ,10
aThe pBAC-V2 clone was used as the HSV-1 helper construct in all
trans-fections.
bSerial 10-fold dilutions of the lysates were plated on a confluent lawn of Vero
cells, and the cells were assayed forb-galactosidase activity after 2 days by using X-Gal histochemistry.
cSerial 10-fold dilutions of the lysates were plated on a confluent lawn of Vero
cells, and plaques were counted after 3 days. A titer of,10 reflects the fact that no helper virus was detected in a 1021dilution of the lysate, which was the lowest
dilution tested in this experiment.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.50.290.82.156.2]lian cells. Note, however, that the pBAC-V1 and pBAC-V2 clones described in this study have the a and UL41 sequences
deleted and are thus not well suited for most other studies.
ACKNOWLEDGMENTS
We thank Marilyn McLeod for excellent technical assistance through-out this project, Aly Cassam for providing the differentiated PC12 cells, Jim Smiley for forwarding his protocol for the purification of HSV genomic DNA, and Greg Dekaban, Mick Bhatia, and Grant McFadden for helpful discussions and comments on the manuscript. We are indebted to Jim Smiley, Dora Ho, Cornel Fraefel, Alfred Geller, Andrew Davison, Melvin Simon, Hiroaki Shizuya, Paul John-son, Ted Friedman, Doug Bell, and David Baltimore for their gener-osity in providing the various cell lines and plasmid constructs.
This work was supported by an operating grant to C.A.S. from the Medical Research Council of Canada. C.A.S. is supported by a Cancer Research Society/Medical Research Council scholarship, and T.A.S. is supported by an Ontario Graduate Scholarship.
REFERENCES
1. Breakefield, X. O., and N. A. DeLuca. 1991. Herpes simplex virus for gene delivery to neurons. New Biol. 3:203–218.
2. Chiocca, E. A., B. B. Choi, W. Z. Cai, N. A. DeLuca, P. A. Schaffer, M. DiFiglia, X. O. Breakefield, and R. L. Martuza.1990. Transfer and expres-sion of the lacZ gene in rat brain neurons mediated by herpes simplex virus mutants. New Biol. 2:739–746.
3. Chou, J., and B. Roizman. 1985. Isomerization of herpes simplex virus 1 genome: identification of the cis-acting and recombination sites within the domain of the a sequence. Cell 41:803–811.
4. Cunningham, C., and A. J. Davison. 1993. A cosmid-based system for con-structing mutants of herpes simplex virus type 1. Virology 197:116–124. 5. Fink, D. J., N. A. DeLuca, W. F. Goins, and J. C. Glorioso. 1996. Gene
transfer to neurons using herpes simplex virus-based vectors. Annu. Rev. Neurosci. 19:265–287.
6. Fraefel, C., D. R. Jacoby, C. Lage, H. Hilderbrand, J. Y. Chou, F. W. Alt, X. O. Breakefield, and J. A. Majzoub.1997. Gene transfer into hepatocytes mediated by helper virus-free HSV/AAV hybrid vectors. Mol. Med. 3:813– 825.
7. Fraefel, C., S. Song, F. Lim, P. Lang, L. Yu, Y. Wang, P. Wild, and A. I. Geller.1996. Helper virus-free transfer of herpes simplex virus type 1 plas-mid vectors into neural cells. J. Virol. 70:7190–7197.
8. Geller, A. I., and X. O. Breakefield. 1988. A defective HSV-1 vector ex-presses Escherichia coli beta-galactosidase in cultured peripheral neurons. Science 241:1667–1669.
9. Geller, A. I., K. Keyomarsi, J. Bryan, and A. B. Pardee. 1990. An efficient deletion mutant packaging system for defective herpes simplex virus vectors: potential applications to human gene therapy and neuronal physiology. Proc. Natl. Acad. Sci. USA 87:8950–8954.
10. Glorioso, J. C., W. F. Goins, D. J. Fink, and N. A. DeLuca. 1994. Herpes simplex virus vectors and gene transfer to brain. Dev. Biol. Stand. 82:79–87. 11. Ho, D. 1994. Amplicon-based herpes simplex virus vectors. Methods Cell
Biol. 43:191–210.
12. Ho, D. Y., T. C. Saydam, S. L. Fink, M. S. Lawrence, and R. M. Sapolsky. 1995. Defective herpes simplex virus vectors expressing the rat brain glucose transporter protect cultured neurons from necrotic insults. J. Neurochem. 65:842–850.
13. Huard, J., W. F. Goins, and J. C. Glorioso. 1995. Herpes simplex virus type 1 vector mediated gene transfer to muscle. Gene Ther. 2:385–392. 14. Johnson, P. A., A. Miyanohara, F. Levine, T. Cahill, and T. Friedmann. 1992.
Cytotoxicity of a replication-defective mutant of herpes simplex virus type 1. J. Virol. 66:2952–2965.
15. Johnson, P. A., M. J. Wang, and T. Friedmann. 1994. Improved cell survival by the reduction of immediate-early gene expression in replication-defective mutants of herpes simplex virus type 1 but not by mutation of the virion host shutoff function. J. Virol. 68:6347–6362.
16. Johnson, P. A., K. Yoshida, F. H. Gage, and T. Friedmann. 1992. Effects of gene transfer into cultured CNS neurons with a replication-defective herpes simplex virus type 1 vector. Brain Res. Mol. Brain Res. 12:95–102. 17. Krisky, D. M., P. C. Marconi, T. Oligino, R. J. Rouse, D. J. Fink, and J. C.
Glorioso.1997. Rapid method for construction of recombinant HSV gene transfer vectors. Gene Ther. 4:1120–1125.
18. Lachmann, R. H., and S. Efstathiou. 1997. Utilization of the herpes simplex virus type 1 latency-associated regulatory region to drive stable reporter gene expression in the nervous system. J. Virol. 71:3197–3207.
19. Leib, D. A., and P. D. Olivo. 1993. Gene delivery to neurons: is herpes simplex virus the right tool for the job? Bioessays 15:547–554.
20. Lim, F., D. Hartley, P. Starr, P. Lang, S. Song, L. Yu, Y. Wang, and A. I. Geller.1996. Generation of high-titer defective HSV-1 vectors using an IE 2 deletion mutant and quantitative study of expression in cultured cortical cells. BioTechniques 20:460–469.
21. MacGregor, G. R., A. E. Mogg, J. F. Burke, and C. T. Caskey. 1987. Histo-chemical staining of clonal mammalian cell lines expressing E. coli beta galactosidase indicates heterogeneous expression of the bacterial gene. So-mat. Cell Mol. Genet. 13:253–265.
22. Messerle, M., I. Crnkovic, W. Hammerschmidt, H. Ziegler, and U. H. Kos-zinowski.1997. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 94: 14759–14763.
23. Paterson, T., and R. D. Everett. 1990. A prominent serine-rich region in Vmw175, the major transcriptional regulator protein of herpes simplex virus type 1, is not essential for virus growth in tissue culture. J. Gen. Virol. 71: 1775–1783.
24. Pear, W. S., G. P. Nolan, M. L. Scott, and D. Baltimore. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA 90:8392–8396.
25. Pechan, P. A., M. Fotaki, R. L. Thompson, R. Dunn, M. Chase, E. A. Chiocca, and X. O. Breakefield.1996. A novel ’piggyback’ packaging system for herpes simplex virus amplicon vectors. Hum. Gene Ther. 7:2003–2013. 26. Roizman, B. 1979. The structure and isomerization of herpes simplex virus
genomes. Cell 16:481–494.
27. Roizman, B., A. B. Sears, and R. J. Whitley. 1996. Fields virology, 3rd ed. Lippincott-Raven, New York, N.Y.
28. Samaniego, L. A., L. Neiderhiser, and N. A. DeLuca. 1998. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72:3307–3320.
29. Samaniego, L. A., N. Wu, and N. A. DeLuca. 1997. The herpes simplex virus immediate-early protein ICP0 affects transcription from the viral genome and infected-cell survival in the absence of ICP4 and ICP27. J. Virol. 71: 4614–4625.
30. Shizuya, H., B. Birren, U. J. Kim, V. Mancino, T. Slepak, Y. Tachiiri, and M. Simon.1992. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl. Acad. Sci. USA 89:8794–8797.
31. Smibert, C. A., D. C. Johnson, and J. R. Smiley. 1992. Identification and characterization of the virion-induced host shutoff product of herpes simplex virus gene UL41. J. Gen. Virol. 73:467–470.
32. Smiley, J. R., C. Lavery, and M. Howes. 1992. The herpes simplex virus type 1 (HSV-1) a sequence serves as a cleavage/packaging signal but does not drive recombinational genome isomerization when it is inserted into the HSV-2 genome. J. Virol. 66:7505–7510.
33. Smith, I. L., M. A. Hardwicke, and R. M. Sandri-Goldin. 1992. Evidence that the herpes simplex virus immediate early protein ICP27 acts post-transcrip-tionally during infection to regulate gene expression. Virology 186:74–86. 34. Spaete, R. R., and N. Frenkel. 1985. The herpes simplex virus amplicon:
analyses of cis-acting replication functions. Proc. Natl. Acad. Sci. USA 82: 694–698.
35. Starr, P. A., F. Lim, F. D. Grant, L. Trask, P. Lang, L. Yu, and A. I. Geller. 1996. Long-term persistence of defective HSV-1 vectors in the rat brain is demonstrated by reactivation of vector gene expression. Gene Ther. 3:615– 623.
36. Stevens, J. G. 1989. Human herpesviruses: a consideration of the latent state. Microbiol. Rev. 53:318–332.
37. Strathdee, C. A. 1996. Development of a hybrid herpesvirus vector for gene expression in mammalian tissues, abstr. W31-8. 15th Annual Meeting of the American Society for Virology.
38. Strathdee, C. A. Unpublished data.
39. Tischler, A. S., and L. A. Greene. 1978. Morphologic and cytochemical properties of a clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Lab. Invest. 39:77–89.
40. Tsui, L. C., M. L. Breitman, L. Siminovitch, and M. Buchwald. 1982. Per-sistence of freely replicating SV40 recombinant molecules carrying a select-able marker in permissive simian cells. Cell 30:499–508.
41. Wang, S., and J. M. Vos. 1996. A hybrid herpesvirus infectious vector based on Epstein-Barr virus and herpes simplex virus type 1 for gene transfer into human cells in vitro and in vivo. J. Virol. 70:8422–8430.
42. Wu, N., S. C. Watkins, P. A. Schaffer, and N. A. DeLuca. 1996. Prolonged gene expression and cell survival after infection by a herpes simplex virus mutant defective in the immediate-early genes encoding ICP4, ICP27, and ICP22. J. Virol. 70:6358–6369.
43. Wu, X., Y. Leduc, M. Cynader, and F. Tufaro. 1995. Examination of condi-tions affecting the efficiency of HSV-1 amplicon packaging. J. Virol. Methods 52:219–229.
44. Zhang, X., H. O’Shea, C. Entwisle, M. Boursnell, S. Efstathiou, and S. Inglis.1998. An efficient selection system for packaging herpes simplex virus amplicons. J. Gen. Virol. 79:125–131.