p. 104-112 Vol.
0022-538X/82/070104-09$02.00/0
Site-Specific Phosphorylation
Regulates the Transcriptive
Activity of Vesicular Stomatitis Virus NS Protein
CHUNG-HSIUNG HSU, EXEEN M. MORGAN,ANDD. W.KINGSBURY* Divisionof Virology, St.Jude Children's ResearchHospital,Memphis, Tennessee 38101Received 1 December1981/Accepted25 March 1982
Invitrotranscription by vesicular stomatitis virus nucleocapsids is inhibited by
enzymatic dephosphorylation of the NS protein. We provide evidence that
specific, partial dephosphorylation ofNSmoleculesis theonlydetectablechange
innucleocapsidstreated with bacterial alkalinephosphatase under conditions that
preventthe action of adventitiousprotease.Dephosphorylationappearedto affect
only the rateoftranscription; therewere no changes in sedimentation rates of
transcripts. To identify the sites of phosphorylation required for NS activity in
transcription,weexaminedphosphopeptides produced by chymotrypsin digestion
ofthetwoelectrophoretic classes ofNS molecules found in virions and infected
cells. The electrophoretically slower class, NS1, abundant in the intracellular soluble pool, hasalower activity in transcription; it contained six chymotryptic
phosphopeptides. Five of these peptides contained both phosphoserine and
phosphothreonine, indicating that this peptide cluster represents at least 11
separate sites ofphosphorylation. In theelectrophoretically faster nucleocapsid-associated NS2 class of molecules, which supportahigher rate of transcription, anothergroupofeight phosphopeptideswassuperimposedonthispattern. Two of these peptides contained both phosphoserine and phosphothreonine, so this cluster ofpeptides representsat least 10 additionalphosphorylation sites. These sites were especially sensitive to dephosphorylation bybacterial alkaline phos-phatase. Oneor moreof them appearstoberesponsiblefor thehigher transcrip-tionratesmediatedbyNS2 molecules.
The NSphosphoproteinof vesicular
stomati-tis virus (VSV)participates in viral RNA
synthe-sis(3).Arequirementforhighlevelsof
covalent-ly bound phosphate for full activity of NS in transcription was indicated by the in vitro recon-stitution experiments ofKingsford andEmerson (15). After removing NS molecules from viral nucleocapsids, these authors replaced them with several different classes of NS molecules that differed in extent of phosphorylation; only the most highly phosphorylated NS molecules re-stored full transcriptiveactivity.
Thesefindings were confirmed by Kingsbury et al. (14), who observed inhibition of in vitro transcription by VSVnucleocapsids after diges-tion with bacterial alkaline phosphatase (BAP). In this paper, we document that the enzymatic dephosphorylation of NS is specific under ap-propriate conditions, and we identify two sub-setsof phosphorylatedsites on the NSmolecule that have different effects on its transcriptive function.
MATERIALSANDMETHODS
Virus. Monolayer cultures of BHK cells were infect-edataninput multiplicity of 0.01 PFU per cell. For radiophosphate labeling, the cells were washed with
phosphate-free Eagle minimal essential medium 4 h after infection and then incubated for 16 h in phos-phate-free minimal essential medium containing 100 ,Ci of carrier-free 32Pi per ml and3% dialyzed fetal calfserum. Under theseconditions, most of the cells becameinfected secondarily, providing time for equili-bration of the intracellular ATP pool with radiophos-phate before significant viral protein synthesis began. About70% of the radioactivity added was found in the medium at the end of the incubation, indicating that an adequate supply of extracellular radiophosphate had been maintained.
[35S]methionine labeling was done in the same way, except theconcentration of radioactivity was 30,uCi/ ml and the serum added to the medium was not dialyzed.
Enzymaticdephosphorylation. Virions were disrupt-edwith1% Triton X-100 in 0.15 M NaCI-0.01 M Tris-hydrochloride (pH 8.0) at room temperature. BAP fromEscherichia coli (Worthington Diagnostics,
prod-uct code BAPF; specific activity, 30 to 40 U/mg of protein) was added at the concentrations given in the figure legends, and incubation was performed for 1 h at 30°C. Aprotinin (Sigma Chemical Co.) was added to all BAP preparations in the proportion of 1 part to 10 parts of BAP by weight.
When transcriptase activity was to be measured, BAPwas removed by centrifuging the nucleocapsids
at45,000 rpm (SW55rotor) for 90minat 8°Cthrough a layer of30oglycerol onto a1.33-g/cm3D20-sucrose
104
on November 10, 2019 by guest
http://jvi.asm.org/
NS PHOSPHORYLATION SITES 105
cushion. Transcriptase activity was measured as de-scribed byBaneijeeand Rhodes (1).
Peptide mapping.32P-labeledNS protein was isolat-ed by polyacrylamideslab gel electrophoresis (4). NS was electrophoretically eluted from gel segments in buffer containing 300 ,ug of sodium dodecyl sulfate-denatured ovalbumin per ml, and both proteins were precipitated with 10 volumes of ethanol. The proteins were collected by centrifugation, denatured with gua-nidine-hydrochloride (9), alkylated with iodoaceta-mide (13), dialyzed against 0.025 M NH4HCO3 (pH 8.0), and digested at 37°C with Nab-p-tosyl-L-lysine chloromethyl ketone-treated chymotrypsin (Worthing-ton). The enzyme was added in two portions of 30 ,ug/ ml each, separated by an interval of 3 h, with incuba-tion extended for 16 h after the addiincuba-tion of the second portion. The digest waslyophilized three times from water to remove salt, spotted on a 0.1-mmcellulose F glassthin-layer chromatography plate (E. Merck AG, Darmstadt), and electrophoresed in glacial acetic acid-pyridine-water (1:3:%, pH 5.5) at 1,000 V for 1 h on a coolingplate at 13°C. The plate was dried overnight and chromatographed in N-butanol-glacial acetic acid-pyridine-water (17:12:35:35, pH 4.0). Phospho-peptides werevisualized by autoradiography.
Phosphoamino acid identification. Phosphopeptides identified by autoradiography were scraped from thin-layer chromatography plates and hydrolyzed in 6 N HCIat110°C for 1.5 h. Afterlyophilizationtwice from water, samples were electrophoresed on CEL 400-10 cellulose glass thin-layer chromatography plates (Ma-cherey, Nagel and Co.) at 2,000 V for 1 h in glacial acetic acid-formic acid-water (7.8:2.5:90, pH 1.9). Autoradiography was then performed, and the posi-tions of phosphoamino acids were compared with ninhydrin-stainedmarker phosphoamino acids.
RESULTS
Specific removal of phosphate by BAP. We have presented evidencethat BAP can be used safelytodephosphorylate NS ifa protease that contaminates BAP preparations is inhibited by
thepeptide Aprotinin (14). This is documented
further inFig. 1,whereautoradiography reveals
no breakdown ofany species of virion protein
after incubation with the maximum
concentra-tion of BAP used in our experiments. Also
shown inFig. 1is thestepwise disappearanceof
NS2, the more highly phosphorylated
electro-phoretic form of NS (4, 11), at lower BAP
concentrations and the partialresistanceof the less phosphorylated NS1 species to even the
highestBAPconcentration. Underthese
condi-tions,NS2 iscompletelyconvertedtoNS1.This
cannot be seen in the [35S]methionine-labeled
examples shown in Fig. 1, but it has been
documentedpreviously (11, 14).
Although the covalentintegrityofthe
nucleo-capsid proteins L, N, and NS appeared to be
maintained after
enzymatic
dephosphorylation,it waspossible that removal of
phosphate
fromNSmightsecondarilyalter itsown
affinity
ortheaffinity of the L
protein
for thenucleocapsid.
However, most or all of NS and L remained
p
C 25 50 00
NS1-0*
w*
NS2-:
S
C 100
--L
-"N
SI
IIPIIS
_-M
FIG. 1. Dephosphorylation of VSV NS protein. Virions labeled with
32Pi
or [35S]methionine were disruptedwith Triton X-100 and incubated with BAP inthe presence of Aprotinin as described in the text. Afterincubation at 30°C for 1 h, samples were precip-itated with 5% trichloroacetic acid and subjected to polyacrylamidegel electrophoresis (4). Abbreviations: P, 32P-labeledvirions; S, 35S-labeledvirions; C, con-trol,noBAP; numerals designate micrograms of BAP per 0.25ml.associated with BAP-treated nucleocapsids after centrifugation through 30% glycerol (Fig.2). We have shown elsewhere that the viralRNAwithin nucleocapsids had an unaltered sedimentation rateafterBAP treatment (14).
Residual BAP in the transcriptase assay. Re-moval of BAP from treated nucleocapsids is a prerequisite foraccurateestimation of remaining transcriptase activity, since BAP dephosphory-lates the nucleoside triphosphate substrates of the transcriptase (18). Indeed, we found that
transcription was inhibited about 30% by the
addition of10 ,ugof BAPpermltothe transcrip-tase reaction (0.3 to 0.4 U/ml), and 100 ,ugof BAP perml resulted incompleteinhibition(data not shown). However, BAP concentrations
be-low 1 ,ug/ml were not inhibitory, and direct
spectrophotometricassay(8)revealed that BAP
contaminationwas atleast 10-fold less than this level in nucleocapsids centrifuged through30% glycerol (see above).
Products of the inhibited transcriptase. We reportedthat 70to80% ofthe in vitro
transcrip-tase activity of VSV nucleocapsids is inhibited
byexhaustive BAPtreatment(14).Theproduct
analyses ofFig. 3 show that there is no
differ-encein thesedimentationpropertiesof the RNA
speciesmadeafter BAPdephosphorylation. On
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.504.274.443.53.278.2]106 HSU, MORGAN, AND KINGSBURY
1 2 3
NI
NS
-0A
FIG. 2. Recovery of nucleocapsid proteins after BAP treatment.Virionsweredisruptedwith Triton X-100 and treated with BAP, and nucleocapsids were isolated by centrifugation through 30% glycerol (see text). After electrophoresis (4), the gel was stained with Coomassieblue. Lanes: 1,noBAP, heldat0°C; 2, no BAP, incubatedat30°Cfor1h; 3,400 ,ugof BAP perml, incubatedat30°Cfor 1h. A smallamountof M protein remained attachedtonucleocapsidsisolated in this manner.
the onehand, the paucityofgenome-sizeRNA
moleculesindicates thattranscription,not repli-cation, comprises the residual activity of
de-phosphorylated nucleocapsids. On the other
hand, the absence of smaller RNA molecules indicates thatinhibition of RNAsynthesisisnot
dueto aprematureterminationofnascentRNA chains andsuggeststhat the role ofphosphatein NS protein function is to enhance the rate of transcript initiationorelongationorboth. More discriminating analyses are needed to decide whether transcription of individual genes or post-transcriptional RNA processing is altered by BAP.
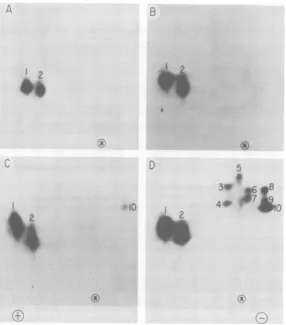
Phosphorylated sites in NS molecules. NS mol-ecules with higherlevels of phosphorylation are more abundant in virions than in infected cells (10), andthey are more active intranscription in vitro(15). To obtain more information about the structural basis of NS phosphorylation and its relationshiptotranscriptase activity, we digest-ed denatured NS molecules with proteolytic enzymes and separated the resulting phospho-peptides by two-dimensional electrophoresis and chromatography. Preliminary trials with trypsin digestion yielded smeared and poorly resolved spots (data not shown). We were more successfulwithchymotrypsin, which gave more discrete products (Fig. 4). As many as 10
phos-phopeptide spots were seen in a digest of the more highly phosphorylated subspecies, NS2 (Fig. 4D). However,twoofthesespots, labeled 1 and 2, weredisproportionately large, indicat-ingthat they represented groups of phosphopep-tides with similar mobilities. Thissuspicionwas confirmed when three subspecieswereresolved in each of these spots on a different cellulose matrix(Fig. 5). The peptides in groups 1 and 2 were present in every sample of NS that we
examined, including NS1 from the soluble pool ofinfected cells (Fig. 4A),NS1 from intracellu-lar nucleocapsids (Fig. 4B), and both electro-phoretic forms of NS from virions(Fig. 4C and D). The ubiquity and relative resistance to de-phosphorylation ofthese phosphopeptides (see
below) led to the designation, primary cluster.
Migration toward the anode and a fairly low mobility in the ascending chromatographic di-mension indicate that this cluster is relatively acidicin netchargeandhydrophilic.
2
0
x
CL
iAJ
I0 I
B D
I0
5
0 10 20 30
FRACTION
10 20 30
FIG. 3. Sedimentation analysis of RNA transcripts made after BAP treatment. VSV nucleocapsids were incubated at 30°C for 2 h in a transcriptasereaction mix containing[3H]UTP(1).Product RNA was isolat-edby phenol-sodium dodecyl sulfate extraction and centrifuged in 15 to
30%o
sucrose gradients. Before centrifugation half of each sample was denatured with glyoxal(16). Panels: (A) RNA made bynucleocapsidsnottreated withBAP; (B) RNA made byBAP-treated nucleocapsids; (C) as (A), but glyoxylated; (D) as(B), but glyoxylated. Vertical arrows represent rRNA markers; these did not separate in glyoxylated sam-ples. Sedimentation is shown from left to right.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.504.115.187.75.269.2] [image:3.504.267.455.302.554.2]NS PHOSPHORYLATION SITES 107
IA
.x
D
2
I0
5
30
.( V
f<i :~~~~~~~~X
FIG. 4. Phosphopeptides in NS molecules.
32Pi-labeled
NS molecules from infected cells and virions were isolatedby gelelectrophoresis and digested with chymotrypsin. The digests were separated by two-dimensional electrophoresis andchromatography in cellulose F thin layers (see text), and autoradiograms were prepared. Panels: (A) NS1 from the soluble intracellular pool; (B) NS1 from free intracellularnucleocapsids;(C)NS1 from virions; (D) NS2 from virions. Symbols:0&
siteof sampleapplication;(®,
anode;E,cathode. Electrophoresis wasin the horizontal dimension.NS1 isthe only form of NS thatweobserved inthe soluble pool; KingsfordandEmerson (15) have shown that NS molecules from thesoluble pool do not restore full transcriptive activity when added to nucleocapsid templates. This
indicates that phosphorylation of primary
clus-tersites doesnotfully activate NS for its role in transcription.
The phosphopeptides in spots 3 to 10 were
characteristicof NS2 molecules (Fig. 4D). This
group of phosphopeptides is relatively basic
andhydrophobic. Thefact that itwas
superim-posedupontheprimarycluster earnedthe desig-nation, secondary cluster. Since molecules with
the electrophoretic mobility of NS2 activate
transcriptionfully(14,15), the sitesresponsible
for thisactivityevidentlyreside inthesecondary cluster.
There were markeddifferences in the intensi-ties of the radioactive spots in Fig. 4 and 5.
Quantitatively, 60% of the total radiophosphate in NS2 molecules was in the primary cluster, and the relative incorporation into individual peptides within this cluster spanned a 13-fold range(peptides la and lc in Table 1). The range of radioactivity in the secondary cluster was about half as great. Since steady-state labeling conditions wereemployed (seeabove),we con-clude that the efficiency ofphosphorylation of different phosphorylation sites ortheirstability varies considerably within both clusters and that themajorityofmolecules,evenin the more highly phosphorylated NS2 class, are incom-pletely phosphorylated.
Phosphoamino acid analyses (Fig. 6) showed
that both phosphothreonine and phosphoserine
were present in most members of the primary cluster(spotlaappearedtolack phosphothreo-nine).In thesecondarycluster, phosphothreon-ine andphosphoserinewere seeninspots3 and
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.504.115.402.71.397.2]108 HSU, MORGAN, AND KINGSBURY
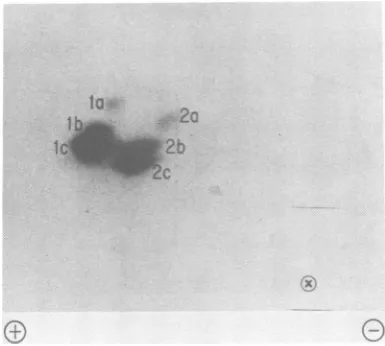
I2a
FIG. 5. Further resolution of phosphopeptides in theprimary cluster.
32P,-labeled
NS1 from virions was examined as described in Fig. 4, but the thin-layer chromatography plate was CEL40010 cellulose.4, but phosphoserine was the only substituted amino acid recoveredfrom theremaining mem-bers of this group. In all, seven peptides con-tained both phosphoamino acids, defining 14 sites of phosphorylation,in addition to the seven sites defined by peptides containing only phos-phoserine. Phosphotyrosine was not observed, in agreement with the findings of Clinton and Huang (5), who recovered only phosphoserine andphosphothreoninefrom NS molecules phos-phorylated in vivo.
...1.7
^.
* *,_J_.'
,
9I
[image:5.504.259.454.74.196.2]~.99e,. ***
TABLE 1. Radiophosphate in NS2chymotryptic peptidesa
Peptideb Relative cpmc Peptideb Relativecpmc
la 1.0 7 2.0
6 1.1 3 2.8
8 1.2 2b 3.9
2a 1.3 lb 4.3
4 1.4 2c 5.7
5 1.4 10 7.0
9 1.7 lc 13.0
aRadioactive spots were located on thin-layer chromatography peptide maps by autoradiography, scrapedfrom the backing, and counted in PCS solubi-lizer.
b Phosphopeptide spots are listed in order of in-creasing radioactivity.
cAverage of duplicate determinations.
BAP-labile phosphopeptides. As reported pre-viously (14) and as shown in Fig. 1, NS2 was more sensitive to BAP than was NS1. After moderate BAP treatment of virion nucleocap-sids containing both NS1 and NS2, NS mole-cules migrating in the NS1 positionweredevoid of phosphopeptides in the secondary cluster (Fig. 7). This confirms the role of the secondary clusterofphosphorylation sites in increasing the electrophoretic mobility of the protein. Exhaus-tive treatment with BAP eventually resulted in dephosphorylation within the primary cluster, especially in group 1 phosphopeptides (Fig. 7B). More work will be needed to determine the relative importance of BAP-sensitive and
BAP-I
g e
f,
I
FIG. 6. Phosphoamino acids in NS chymotryptic peptides. Thin-layer electrophoresis was performed after acid hydrolysis (see text) of32P-labeledpeptides isolated from two-dimensional maps. The number at the head of each lane designates peptide spots identified in Fig. 4. Abbreviations: P04,
P,;
P-Ser,phosphoserine;
P-Thr,
phosphothreonine.Incomplete hydrolysis left some slowly migrating radioactivity near the origin.
VIROL.
.".. .,:
I .. t..
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.504.54.247.76.249.2] [image:5.504.111.396.439.631.2]NS PHOSPHORYLATION SITES 109
la
lb
2o
IC
-2b
2c
A
.qiiiiiii.
B
FIG. 7. Phosphopeptides remaining after BAP treatment.Nucleocapsids from 100
pg
of virionswere treatedwith BAP for 1 h at 30°C in a volume of 0.5 ml as described in the text. After treatment, only NS1 molecules were seen on polyacrylamide gel electro-phoresis. Panels: (A) 100Fg of BAP; (B) 400p.g of BAP.resistant sites within the primary cluster for transcriptive activity, but it is clear that a base-line level of activity representing about 20%oof maximum (14) remains when dephosphorylation is as severe asshown in Fig. 7B.
Superphosphorylation
andrephosphorylation.
VSvirionscontain a protein kinase that is
capa-bleofsuperphosphorylating NS as well as virion
envelope protein M (12, 17). This kinase
ap-pearedtohaveamarked preference for sites in
theprimarycluster,judging by the
electropho-reticandchromatographic behavior of the
phos-phopeptides resulting frominvitro
phosphoryla-tion (Fig. 8). In addition, the small amount of
radiophosphate
appearing
in therelativelybasicandhydrophobicpeptidescorresponded poorly
to thepattern of the secondary cluster seen in
moleculesphosphorylated in vivo (Fig.4). Allof
these results indicate that thevirion kinase has
proteinsubstrate specificitiesdifferent from the
intracellularkinases thatphosphorylateNS
mol-ecules.
Rephosphorylation of BAP-treated NS
mole-cules was minimal and restrictedto two
phos-phopeptides ofahydrophobicandbasic
charac-ter(Fig. 8C).Extensive
dephosphorylation
maychangetheconformation of NS molecules
mark-edly, reducing the availability of sites to the
virion kinase. Alternatively,the residual
protein
B
(6) (8) (12) (10
C
(9)
09
0
FIG. 8. Superphosphorylation and
rephosphoryla-tion of NSprotein.Numbers inparenthesesrepresent tentative peptide assignments (Fig. 4). Virions were disruptedwithTritonX-100andwerephosphorylated invitrobyendogenousproteinkinase(14).Panels:(A) NS1; (B) NS2(theinset isashorter exposureof the
primary clusterofphosphopeptides); (C)after exten-sivedephosphorylation with BAP(Fig. 7B), nucleo-capsidswereisolatedby centrifugationthrough 30%A glycerolandrephosphorylatedin vitrobytheprotein kinase thataccompaniedthem(14).Theradioactivity migratedonelectrophoresisasNS1.
VOL.43, 1982
(11)
..v.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.504.85.231.72.302.2] [image:6.504.287.434.143.563.2]HSU,
kinase that remained attached to isolated nu-cleocapsids may have been insufficient to re-phosphorylate effectively. In either case, the limited amount ofphosphorylation obtained is
insufficient to restoretheelectrophoretic
migra-tion of these molecules to the NS2 position, confirming that rephosphorylation during the transcriptase assay is too limited to restore
function (14).
DISCUSSION
We have presented evidence that the NS protein of VSVcanbedephosphorylated specifi-cally by BAP. However, our data showing no
changes inthe electrophoretic migration of the
viral proteins (Fig. 1) or in the sedimentation
rate of virion RNA (14) do notrule out subtle
changes causedbycontaminatingenzyme
activi-tiesinourBAPpreparation. Wecouldnothave
detected limitedcleavageof terminal amino ac-ids from any of the nucleocapsid proteins or removalof terminal nucleotides from the virus genome. Even limitedchangeslike these might have severe functional consequences, so until these possibilities have been checked, there mustbeanelement of doubt about the functional significance ofourenzymaticdephosphorylation results.Nevertheless,ourconclusionsabout the
functional relationships ofphosphorylated sites
in NS1 and NS2 molecules donotdepend entire-lyonourworkwith BAP;theyare independent-ly supported by the experiments ofKingsford
andEmerson(15),who showeddirectlythat the
more highly phosphorylated molecules of the NS2 class conferred a higher rate of transcrip-tion on VSVnucleocapsids than did molecules of the NS1 class.
NS is anexceptional protein in the extent of
phosphorylation that it sustains. Generally,
phosphoprotein enzymes that are regulated by
reversible phosphorylationhave only one or two phosphorylated sites (6). The primary sequence of NSindicatesthepresence of 12threonineand 21 serine residues, a total of 33 potential sites of phosphorylation (7). The phosphorylation of mostof these sites is realized in vivo, since we resolved14phosphopeptidesafterchymotrypsin
digestionand7 of these peptides contained both
phosphoserine and phosphothreonine, identify-ing at least 21 phosphorylated sites. Further proteolytic digestion of NS may well uncover
additionalsiteswithin thechymotrypticpeptides
that we resolved.
Ourchymotryptic peptide maps indicate that there are two classes ofphosphorylated sites in NS that define the two major electrophoretic and functional classes of NS molecules previ-ously described (4, 11, 15). NS1, which is found
mainly in the soluble pool of the cell and in intracellularnucleocapsids,isphosphorylatedin
aprimarycluster of sixphosphopeptides and is
notfullyactive intranscription(15). NS2,found mainlyinvirions,isphosphorylatedbothin the primary cluster of sites and in a secondary cluster of eight phosphopeptides, giving this class ofmolecules more rapid electrophoretic migration and greater activity in transcription. Both electrophoretic classes areheterogeneous collections of variously phosphorylated mole-cules, asshownbythestoichiometriesof radio-phosphateinindividual phosphopeptides(Table 1)andbytheisoelectricfocusingand chromato-graphic separations of NS subspecies within these majorclasses(11, 15).
The presenceofthe primaryphosphopeptide clusterin allformsofNSsuggeststhat it repre-sentsabaseline level ofphosphorylation thatis essential for NS function, although it is not sufficientforfull activity ofthe proteinin tran-scription. Whyshould all of the NS molecules in the soluble pooland manyof the NSmolecules innucleocapsids be limitedtothisprimarylevel of phosphorylation? A possible explanation is that aprotein kinase with specificity limited to the primary cluster is the only kinase thatNS encounters within the solublepoolor in nucleo-capsids thatarefar from the cellular membrane, where virion budding occurs. Secondary site phosphorylation may be mediated by a mem-brane-orcytoskeleton-associated proteinkinase with adifferent specificity. Another factorthat may preventkinaseactiononfree NS molecules is theassociation of theprotein with nucleocap-sids. Binding to nucleocapsids may alter the conformation of NSmolecules, rendering previ-ously hidden sites availablefor kinase modifica-tion.
Speculations about the regulatory significance of NSphosphorylation have focusedon qualita-tive mechanisms, such asswitchingthe synthe-sis ofRNAfrom transcriptiontoreplication.For example, Clintonetal.(3)madeobservationson
thedistributionof electrophoreticspecies of NS
in infected cells, suggesting that more highly phosphorylatedNSmoleculeswereless likely to bind to nucleocapsids and that the binding of lessphosphorylatedNS moleculesincreasedthe rate of RNA replication versus transcription. Independently, Testa and co-workers (20) ar-rivedatasimilarhypothesis; they suggested that dephosphorylation ofanucleocapsid-associated protein (presumably NS) favors replication, basedon results of in vitrotranscription experi-mentswithanATP analog that cannot act as a
y-phosphatedonor. Our in vitro studies provide no support for these ideas, since we observed no
increasesinthe sizes of RNA products made by
partiallydephosphorylated VSV nucleocapsids.
on November 10, 2019 by guest
http://jvi.asm.org/
Alternatively, phosphorylation might regulate transcription exclusively, in a quantitative way. As we saw in our in vitro experiments, nucleo-capsids containing dephosphorylated NS exhib-ited significant levels of residual transcriptive activity. Such levels might be appropriate for intracellular secondary transcription, which mustbe balanced against replication, but subop-timal for primary transcription, which is a pre-requisite for gene expression at the beginning of infection by a negative-strand RNA virus. In-creased phosphorylation of NS molecules in nucleocapsids destined for virions would ensure that infecting nucleocapsids transcribe at the
highest possible rate. This hypothesis can be
tested by comparing the relative transcriptive activities of infecting nucleocapsids and progeny nucleocapsids within the infected cell.
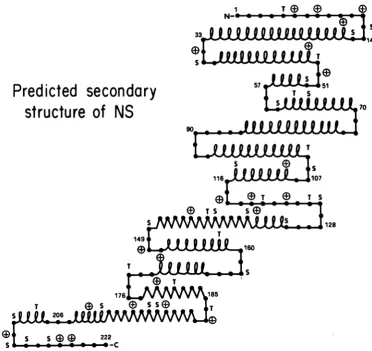
The predicted secondary structure of NS
sug-gests that many potential phosphorylation sites are located at 13-turns that also possess basic amino acid residues (Fig. 9). These are structur-al features that characterize externally located sites of regulatory phosphorylation in other proteins (19). Therefore, we expect to find sec-ondary cluster sites at such 1-turns when we determine the locations of the relevant phospho-peptides in the primary sequence of NS. We also plan to continue to probe the relationships of phosphorylation sites to NS protein function by examining the transcriptive activity of nucleo-capsids treated with lesser amounts of BAP. This may reveal whether any of the secondary clusterof sites aredispensable for full NS activi-ty and whether there is a hierarchy of sites within the secondary cluster that determines accessibility to dephosphorylation and levels of transcriptase activity.
s
Predicted
secondary
structure
of NS
149t
T
I128
T
ED
S W) 5 b ,.T [image:8.504.71.444.288.639.2]20
FIG. 9. Secondarystructureof the NSproteinaspredictedby the method of Chou and Fasman (2). Symbols: AM,a-helix;%M,13-strand;-* * ,random coil; D,abasic amino acid within four residues of a threonine
orserine.1-Turnsareindicatedbychain reversals. Numerals refer to amino acid residues (7). Abbreviations:
S,
serine; T, threonine.
on November 10, 2019 by guest
http://jvi.asm.org/
ACKNOWLEDGMENTS
Paula Pilgrimand Karen Rakestrawprovidedskillful techni-calassistance.WethankJeffrey Siegelfor his Chou-Fasman computer program.
Thiswork wassupported byPublic Health Servicegrants AI05343and CA21765 from the National Institutes of Health and byAmericanLebaneseSyrian AssociatedCharities.
LITERATURE CITED
1. Banerjee, A. K., and D. P. Rhodes.1973. Invitrosynthesis ofRNA thatcontains polyadenylate byvirion-associated RNApolymerase of vesicular stomatitisvirus. Proc. Nati. Acad. Sci. U.S.A. 70:3566-3570.
2. Chou, P. Y., and G. D. Fasman. 1978.Prediction ofthe secondary structure ofproteins from their amino acid sequence.Adv. Enzymol.47:45-148.
3. Clinton, G. M., B. W. Burge, and A. S. Huang. 1978.
Effects of phosphorylationandpHontheassociationof NSprotein with vesicularstomatitisvirus cores. J. Virol. 27:340-346.
4. Clinton, G. M., B. W. Burge, and A. S. Huang. 1979.
Phosphoproteins of vesicular stomatitis virus: identity
andinterconversion of phosphorylated forms. Virology
99:84-94.
5. CUnton, G. M., andA. S. Huang. 1981. Distributionof
phosphoserine,phosphothreonineandphosphotyrosinein proteins of vesicularstomatitis virus.Virology 108:510-514.
6. Cohen, P. 1980. Well established systems of enzyme regulation byreversible phosphorylation, p. 1-10. In P.
Cohen (ed.), Recently discovered systems of enzyme regulation by reversible phosphorylation. Molecular
as-pects of cellularregulation, vol. 1. Elsevier/North Hol-land,Amsterdam.
7. Gallione, C. J., J. R. Greene,L. E. Iverson,and J.K. Rose. 1981.Nucleotide sequences of the mRNA's encod-ingthevesicularstomatitis virus N and NSproteins.J. Virol.39:529-535.
8. Garen, A., and C. Levinthal. 1960. Afine-structure genet-ic and chemgenet-ical study of the enzyme alkaline phosphatase of E. coli.I. Purification and characterization of alkaline phosphatase. Biochim. Biophys. Acta 38:470-483. 9. Gracy, R. W. 1977. Two-dimensional thin-layer methods.
Methods Enzymol. 47:195-204.
10. Hsu, C.-H., and D. W. Kingsbury. 1980. Vesicular
stoma-titis virus morphogenesis is accompanied by covalent protein modifications, p. 613-622. In B. N. Fields, R.
Jaenisch, and C. F. Fox (ed.), Animal virus genetics, ICN-UCLA Symposia on Molecular and CellularBiology, vol. 18. Academic Press, Inc., New York.
11. Hsu, C.-H., and D. W. Kingsbury.1982.NS phosphopro-teinof vesicular stomatitis virus: subspecies separated by electrophoresisandisoelectricfocusing. J. Virol. 42:342-345.
12. Imblum, R. L., and R. R. Wagner. 1974.Protein kinase
and phosphoproteins of vesicular stomatitis virus. J.
Virol. 13:113-124.
13. Jacobson, M. F., J. Asso, and D.Balthnore. 1970.Further evidence on the formation ofpoliovirus proteins.J. Mol.
Biol.49:657-669.
14. Kingsbury, D. W., C.-H. Hsu, and E. M. Morgan. 1981. A rolefor NS-proteinphosphorylation in vesicular stomati-tisvirus transcription, p. 821-827. In D. H. L. Bishop and R. W.Compans(ed.), The replication of negative strand viruses. Developments in cell biology, vol. 7. Elsevier/ NorthHolland, New York.
15. Kingsford, L., and S. U. Emerson. 1980. Transcriptional
activities of differentphosphorylated species of NS pro-tein purified from vesicular stomatitis virions and cyto-plasm of infected cells. J. Virol. 33:1097-1105. 16. McMaster, G. K., and G. G.Carmichael. 1977. Analysis
ofsingle- and double-stranded nucleic acids on polyacryl-amide and agarose gels by using glyoxal and acridine orange. Proc. Natl.Acad. Sci. U.S.A. 74:4835-4838. 17. Moyer, S. A., and D. F. Summers. 1974. Phosphorylation
ofvesicular stomatitis virus in vivo and in vitro. J. Virol. 13:455-465.
18. Reid, T. W., and I. B. Wilson. 1971. E. coli alkaline
phosphatase, p. 373-415. In P. D. Boyer (ed.), The enzymes, vol. 4, 3rd ed. Academic Press, Inc., New York.
19. Smith, J. A., and L. G. Pease. 1980. Reverseturns in
peptidesandproteins. CRC Crit.Rev.Biochem. 8:315-399.
20. Testa, D., P. K. Chanda, and A. K. Banerjee. 1980. In vitro synthesis of the full-length complement of the negative-strand genome RNAofvesicular stomatitis virus. Proc. Natl. Acad. Sci. U.S.A. 77:294-298.
on November 10, 2019 by guest
http://jvi.asm.org/