0022-538X/78/0027-0560$02.00/0
Copyright©) 1978 AmericanSocietyforMicrobiology Printed in U.S.A.
Rebinding of Transcriptase Components (L and NS Proteins)
to
the
Nucleocapsid Template of Vesicular Stomatitis Virus
MARGARET G. MELLON AND SUZANNE U. EMERSON*
University of Virginia Medical School, Department of Microbiology, Charlottesville, Virginia 22901
Received forpublication 7 April 1978
The L and NS proteins of vesicular stomatitis virions (New Jersey serotype) were solubilized with Triton X-100 and high-salt buffer and recombined with purified nucleocapsids under conditions similar to those used to reconstitute transcriptase activityinvitro. Thenucleocapsid-boundL andNS proteins were separated from unbound proteinsonaglycerolgradient. Therebinding of L and NS proteins mimics the invivo binding in that at saturation the ratio of L and NS moleculestoNmoleculesisapproximately the same as observed in the intact
virion. L andNSproteins were separated and added back independently and in
combinationtothetemplate.Thepurified NS proteinbound to thetemplate in the absenceof Lprotein. However,the Lproteinbinding appearedtodepend on
the presence of NS protein. The presence of
Mg2"
and nucleotides, which isrequired for transcription, was not necessary for the rebinding of L and NS proteins.
Most, ifnotall,rhabdoviruses carryan
RNA-dependent RNApolymerase in the virion (14).
In several rhabdoviruses, including the
well-characterizedvesicular stomatitis(VS) virus,the
polymeraseactivity may be dissociated from the ribonucleocapsidtemplateand thetranscription
systemthen reconstitutedbyrecombination of
the separated components (7, 5). Under this reconstitution system, three of the five viral
proteins have been identified as necessary for
transcription (8, 9, 11). These are the major
structural protein Nand the minorproteins L
andNS;thetwomembrane-associatedproteins,
G andM,areapparentlynotrequired (7,6). The
N protein is probably required as an integral
structural component of thetemplate; onlythe
nucleocapsid consistingof Nprotein tightly
com-plexedtotheviralRNAserves as atemplatefor transcription (7, 11).Deproteinizedviral RNA is
not transcribed in the in vitro system. The L
and NSproteins,which have beenpurifiedas a
complex, active in in vitro transcription (11), almostcertainly constitute thetranscriptase
en-zyme. However, mRNAsynthesis catalyzed by
the transcriptase is a complex process. The
mRNA's transcribed in thereconstituted
tran-scription systemarecappedandmethylatedat
their 5' endsandpolyadenylatedattheir 3' ends
(1). Thecappingandpolyadenylating activities
whichmodify the mRNA'sare sotightly coupled
with thetranscriptase activitythat methods for
separating these activities from transcription
haveso fareluded investigators. Thus, the
de-tailedfunctionsofthe L and NSproteinsin the
overall production of mRNA's remain obscure.
As anapproach to understanding the roles of
the L and NS proteins, we have sought
infor-mationontherebindingof thesetwoproteins to
thenucleocapsid of VS virus. In this report we
havedeveloped an assay for the rebinding of L
and NS proteins of VS virus New Jersey to
nucleocapsids and used it to characterize the
bindingcapabilities of the separated L and NS
proteins and to determine the stoichiometry of
and therequirements for the binding reaction.
Since the polymerase rebinds to the template
during transcription, our assay conditions are
basedonthose forthe in vitro reconstitution of
transcriptase activity (7).
MATERIALS AND METHODS
Viruses andcells. TheNewJerseyserotypeofVS
virus wasoriginally obtainedfrom the U.S. Depart-mentofAgriculture, Beltsville,Md., by R. R. Wagner and wasrecentlyclassifiedasthe Concansubtype (13). The viruswasgrownat31°Cinconfluent monolayers of BHK-21 cells in 150-mm2 Corning tissue culture flasks with Dulbecco modified Eagle medium, high glucose, and 10% tryptose broth. Virus was radiola-beledby including150juCiof[3S]methionineperml,
100,uCi of3H-labeledL-aminoacid mixture perml,or
50,Ciof[2H]leucineper ml in theinfectingmedium.
Cultureswereinfected at0.5 to 2 PFU percell, and virions wereharvested at16to18hpostinfection. The
overlaymediumwasclarifiedbya10-min
centrifuga-tionat1,000xg,and the virionswerepelleted through
alayerof 50% glycerolonto apadof Flouroinertby centrifugationat80,000xg for90min. The virus band wasremoved from the surface of the Flourinertpad
andlayeredontoapreformedlinear0to40%sucrose 560
on November 10, 2019 by guest
http://jvi.asm.org/
gradient containing 1 MNaCl and 1mM EDTA (7,
10). The band ofB virions wascollected from the
gradient andconcentratedbycentrifugationat80,000 x g for 90 min. The virus was then centrifuged to
equilibriumovernightonapreformedlinear 10 to40%
sucrose gradient containing 1 M NaCl and 1 mM EDTAandconcentratedagain bycentrifugation.The finalpellet,the harvest of 15bottlesof tissue culture cells (5 to 10mg),wasresuspendedin 1 to 2 ml of 0.01 MTris-hydrochloride (pH7.4) plus15%glyceroland stored at-18°C untilneeded.
Solubilization of viralproteins for rebinding.
Viral proteins for rebinding were labeled with
[3S]-methionine or a 3H-labeled mixture of amino acids. The virus harvest fromapproximately five bottlesof BHK-21cells (-3mgofpurifiedvirus in 0.3ml)was mixed withanequal volume of 2x Triton-high-salt solubilizer(2xHSS=18.7%glycerol,1.44MNaCl,1.2 x 10-3Mdithiothreitol,3.74%Triton X-100, and10-2
M Tris-hydrochloride, pH 7.4) (7). The solubilized virions were incubated at31°C for 30min.The
nu-cleocapsidtemplatewaspelletedfrom thesolutionby centrifugation at 125,000 x gfor 2 h, leaving the
supernatantfractioncontainingthesolubilizedG, M, L, andNS viralproteins. Viralproteinswerealways solubilized immediately before their use in binding experiments.
Templatepurification.Tocompletelyremovethe
tightly adhering NS protein, very dilute samples of virus were used to prepare anucleocapsid template.A
15-bottleharvestofpurifiedvirus(5to10mg) labeled
with[3H]leucineor[3S]methioninewasdilutedto43
ml with 0.01 MTris-hydrochloride, pH7.4.The virions weredisrupted by mixingwith anequal volumeof 2x
HSS (seeabove).Thenucleocapsidswerecentrifuged
onto a pad of renografin at 90,000 x governight,
collectedfrom the surface of thepad,andthen
chro-matographed on anagarose (Bio-GelA-5M) column equilibratedwith 10%glycerol,1MNaCl,0.1%Triton,
10-4Mdithiothreitol, and 0.01 MTris-hydrochloride,
pH 7.4. Approximately2-ml fractionswerecollected
fromthe column. The fractionscontainingthelabeled
nucleocapsids, which eluted with the void volume,
werepooled andcentrifugedthroughalayerof 20%
glycerolonto a100%glycerol padbycentrifugationat 125,000 x g. Thenucleocapsidswere removedalong
with the 100%glycerolpadand storedat-18°Cuntil used.
Binding assays.Details of eachbindingassayare
givenin thefigure legends.The standard assay, based ontheoptimalconditions fortranscription,was setup
accordingtothefollowingscheme (7). Since the salt
optimumfortranscription is -0.15 MNaCl,the viral
proteins, solubilizedin 0.72 MNaCl,werediluted1:4 into thestandardassay mixture togiveafinal NaCl concentration of0.144M(80
id
ofsolubilized proteinsina0.4-ml final reactionvolume).Theprereactionmix (1.4x10-3MATP,GTP, and CTP; 8x10-3
MMg2m
; 1.2x 10-3Mdithiothreitol; and 5x 102MTris, pH 7.4) was two times the desired concentration of nu-cleotides andMg2e,
and 0.2mlof theprereactionmix wasincluded in the 0.4-ml final volume. Various vol-umesofpurifiedtemplate (in100%glycerol) and0.01 MTris-hydrochloride, pH7.4, made up the remaining 0.12ml of the final reaction volume.Glycerol gradients. Linear 15 to 35% glycerol
gradients (5ml)wereformedby usinga custom gra-dient maker. The 15 and 35%glycerol solutionswere made bydiluting either 3 or 7 ml ofglycerolto 20ml with glycerol gradient mix (7 x 10' M ATP, CTP, andGTP, 4 x 10-3 Mmagnesium acetate, 0.144 M NaCl, 2%Triton, and 6 x10-4M DTT in 0.01 MTris, pH 7.4). ATP, CTP, andGTP were included in gra-dientsonly in early experiments. Once it was deter-mined thatnucleotideswerenotrequired for binding, they were omitted from the gradients.
SDS-polyacrylamide gel electrophoresis. Pro-tein sampleswere mixed with 100,ug ofhemoglobin
carrierand 1 volume of 40%trichloroaceticacid and centrifuged at 2,000 x g to pellet the precipitated
protein.Thepellet was washed one or twotimeswith acetoneand dissolved in asolution of 0.01 M
Tris-hydrochloride with 8 M urea, 1% sodium dodecyl
sulfate (SDS),and 1%mercaptoethanol.Afterboiling
for 2min,thesamplesweresubjectedto electropho-resis on 7.5% SDS-phosphate acrylamide gels. Gel
sliceswereincubated with 0.5 ml ofNuclear-Chicago solubilizer(NCS)for 2 h at 50°C and counted in LSC
Complete toluene-based scintillationfluid.
Preparation andrecombination ofthe Land NSprotein fractions. The preparation of L and NS
proteins from VS New Jersey serotype virus is a modification of thepublishedprocedure using VS In-diana serotypevirus(8, 9).Approximately 1 ml (-10 mg) of VS NewJerseyviruslabeledwith [3S]methi-onine wasdilutedto 10ml and mixed with an equal
volume of 2x HSS (see above). The nucleocapsids were removedbycentrifugationfor 2 hat 125,000 x g. The supernatant wasmixedwith 5 to 10 ml of
phos-phocellulose previously equilibratedwith Tris, pH 7.2,
and thendialyzed overnight against column buffer(75 ml ofglycerol,3ml of 20% Triton, 9 mg of DTT, and 222 ml of 5 x 10-2 M Tris-hydrochloride, pH 7.2)
containing0.1MNaCl. The phosphocelluloseand 3S-labeledsupernatantprotein mixturewas then trans-ferred to a 12-mlcolumn, drained,and washed with more0.1M NaCl columnbuffer;theflow-throughwas saved and used later. Thephosphocellulose column waselutedwith 0.5 M NaClcolumn buffer, andthe
peakfractionscontainingthe3S-labeled proteinswere calledthe L-protein fraction. Becauseof the instability
ofthe L protein, the L-protein fractionwas always
usedimmediately.
Thereservedflow-throughfrom the
phosphocellu-losecolumn, containingprimarilyNS andGproteins, wasappliedto a 4- to6-mlDEAE-cellulose column,
and thecolumnwaswashed with 0.1 M NaCl column buffer. The NS proteinwasthen eluted with 0.4 M NaCl in column buffer. Tocompletelyremove L
pro-tein,it wasnecessary todialyzetheNS-protein frac-tionovernight against 0.1 M NaCl column buffer and toprocess the NS proteinthrough anothercycle of
phosphocellulose and DEAE-cellulose chromatogra-phy. Purified NS protein was stored at-180C.
When used inbindingassays, the35S-labeledL-and
NS-proteinfractions were recombinedwith3H-labeled
purified template. The binding assay mixture with both L andNSproteinscontained0.06ml ofL-protein fraction, 0.08ml ofNS-protein fraction,0.025 ml of
template,0.035ml of0.01MTris,pH7.4, and0.2ml
on November 10, 2019 by guest
http://jvi.asm.org/
562 MELLON AND EMERSON
ofprereaction mix(see above), pH7.4. When L- or
NS-protein fractionwasaddedseparately,either 0.08
ml of 0.4 M NaClcolumn wash or0.06ml of0.5M NaCl columnwash, respectively, wasaddedto main-tain thepropersaltconcentration. Thebinding reac-tions wereincubatedat31°Cfor30min.The
template-boundproteinsineachpreparationwereisolatedon
glycerol gradients and analyzed on
SDS-polyacryl-amidegels.
Stoichiometry ofLandNSproteins boundto
template.The relativedistribution of label inL, NS,
and N proteins was obtained from
SDS-polyacryl-amide gels. Smallsamples of intact viruswere dis-solvedingelsamplebuffer andapplied directlytothe
gel.Templateand reboundproteinswereisolatedon
glycerol gradients, trichloroacetic acid precipitated,
and thenanalyzedongels. In the casewhere intact virionswerecomparedtothedissociated and
imme-diately recombined supernatant and pellet fraction,
the virus was labeled with a 3H-labeled mixture of
amino acidstouniformlylabel all viralproteins.The relative amount of radioactivity in L, NS, and N proteinswasdetermined and dividedbythemolecular weightsforL,NS,and Nproteins(190,000,40,000,and 50,000,respectively)togivemolecular ratios. The
mo-lecular ratio for N was divided into 2,000, and the molecular ratiosfor L andNSproteinsweremultiplied
by this conversion factor. The final valuesrepresent the number of L andNS moleculesper2,000N mole-culesonthegel.
Whenpurifiedtemplate andsolubilized viral pro-teinswererecombined,thetemplatewaslabeledwith
[35S]methionine, and the solubilized viral proteins
werelabeled witha3H-labeledmixtureof aminoacids. In thiscasethespecificactivities of the3H-solubilized proteins and the3S-labeledNproteinwerecalculated based on a Lowry protein assay, and the specific
activities of the three proteins were then used to
determine thestoichiometryof L and NSproteinsper
2,000Nmolecules.
Chemicals. [3H]leucineand[3S]methioninewere
obtained fromAmersham/Searle, ArlingtonHeights,
Ill.,and the3H-labeledL-aminoacid mixturewasfrom
NewEnglandNuclearCorp.,Boston,Mass.Whatman
P1l-cellulose and microgranular DEAE-cellulose
DE52 were purchased from Whatman Biochemicals
(Maidstone, England).Renografinwasobtained from
E.R.Squibb &Sons, Princeton,N.J.,and Flouroinert was from Instrumentation Specialties Co., Lincoln,
Neb. LSCCompletescintillation fluidwasfrom York-townResearch,Hackensack,N.J.Agarose(Bio-Gel A-5M), acrylamide, and N,N-methylenebisacrylamide
wereobtained from Bio-RadLaboratories,Richmond,
Calif.
RESULTS
Solubilizedviral proteinsrebound to the template. The L and NS proteins were
solubi-lized along withthe M and Gproteins by treating
whole 35S-labeled virus with Triton X-100 and
high salt (0.72 M NaCl) andpelleting out the
nucleocapsids. This supernatant fraction was used immediately in binding assays. Purified template, consisting ofonlythe N protein and
viral RNA,waspreparedfrom anotherbatch of
3H-labeled virus by standard methods (see
Ma-terialsandMethods).
The purified 3H-labeled template was
recom-binedwith the3S-labeled supernatant proteins
under transcription assay conditions. Final
con-centrationsofMg2+, NaCl, ATP, GTP, and CTP
were optimal for transcription, but UTP was
omittedtopreventtranscription from occurring
(8). Thereaction was incubated at 31°C for 30
min, and then the entire 0.4-mlreactionmixture
was layered on a 15 to 35% glycerol gradient
made up in a solution similar to the reaction
mixture. Figure 1 shows the distribution of
'S
and 3Hlabel inthe glycerol gradient. The
tem-plate along withbound 3S-labeled proteins
sed-imented to the middle of the gradient. The
un-boundproteins remained at the top. Aggregated
template (30 to 50% of total)and proteins(-10%
oftotal) sedimented through the gradient and
pelleted. When supernatant proteins were
ap-plied to thegradient in the absence of template,
no proteins were found in the middle of the
300 200 100
4-V
2 4 6 10
GRADIENT FRACTION
FIG. 1. Glycerol gradient of the binding reaction mixture. High-salt-solubilized VS New Jersey viral proteins labeledwith[3SJmethionine andtemplate labeled with[3H]leucinewereprepared as described in the text. The binding reaction mixture contained 80ulof the solubilizedmS-labeledviralproteins, 70
,ulof template,50,tlof0.1MTris,pH 7.4, and 0.2 ml
ofprereaction mixture (1.4 x 103 MATP, CTP, and
GTP, 1.2x 103 M dithiothreitol, and5 x 102 M
This-hydrochloride,pH7.4).After a30-min
incuba-tionat31°C, the entire0.4-mlreaction mixture was
layeredona 15 to35% glycerol gradient (see text) and
centrifugedat125,000 x g for I h at 4°C. Portions (50
[image:3.501.269.452.321.533.2]1d) of0.5-ml gradient fractions were counted and
plotted. Thedirection of sedimentation is from right toleft.
on November 10, 2019 by guest
http://jvi.asm.org/
gradient(datanotshown).
To determine which of the four solubilized proteins had boundtothetemplate,the
gradient
fractions containingthecosedimenting S- and 3H-labeled proteins were precipitated withtri-chloroacetic acid andanalyzedon an
SDS-poly-acrylamide gel. Despite the fact that M and G proteins were present in much greater abun-dancethanthe LandNS proteins inthe solu-bilized supernatant fraction, only traces of M and Gproteinswerefound
cosedimenting
with template (Fig. 2).Essentially onlythe L and NS proteins reboundtotemplate.Invitro
rebinding
of L and NS proteins mimicked the stoichiometry of intactvin-ons. Inview of the marked
tendency
of the Lprotein toaggregate invitro,it is
important
toshow that the observed cosedimentation of L and NS proteins with template represents a rebinding ofLandNSproteinsto
specific
sites on the template. If the L and NSproteins
arerebinding to
specific
sites on thetemplate,
it 32k
281-
241-x
C4
E
U,1
201-16
[-12H
8
4
AND
should be possible to saturate the available sites
on thetemplate toapproximatelythesame
ex-tent asinundisruptedvirions.
Aseries of binding reactions was set up with
constant amounts of 3S-labeled supernatant proteinsand decreasingamounts of3H-labeled template, in effect increasing the concentration
of Land NSproteins relative to the template.
Each ofthe reactionmixtures was applied to a
glycerolgradient, and the template-bound
pro-teins were analyzed on SDS-polyacrylamide
gels.The amountsof L and NS proteins bound
were normalized to the N protein on the gel.
The amounts ofboundL and NS proteins
in-creasedlinearly with increasing relative
concen-trationsof LandNSproteins and then reached
a plateau, showing that sites on the template
canbesaturatedwith L and NS proteins (Fig.
3).
Further evidence that the binding is
signifi-cant wasobtainedbycomparingthe number of
Land NS proteinsreboundtothetemplateand
N
NS
_ 10 20 30 40 50 +
GEL FRACTION
FIG. 2. SDS-polyacrylamidegelelectrophoresisof template-boundproteins.Fractions4,5, and6fromthe
gradientrepresented inFig.)werepooled,and theproteinswereanalyzedonSDS-phosphatepolyacrylamide
gels.Thetemplatewaslabeled with3H,and the solubilizedsupernatantproteinswerelabeled with 'S. No
!2M
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.501.108.413.319.639.2]MELLON AND EMERSON
cn
5.
-n
2
0
3
2
1
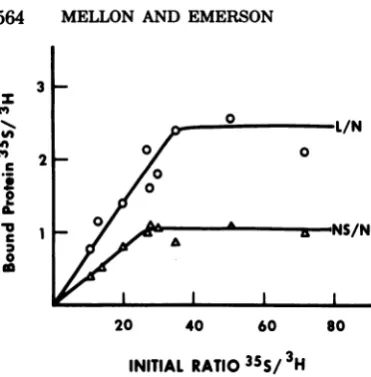
FIG. 3. Bindi A seriesofbind
asdescribed in
mlof theprerea
lized viralprote ,ul) of 3H-label hydrochloride, bring the total boundfromunl
gradients, theI the rebound L SDS-polyacryla
orNSproteins
labeled Nprot plotted against lizedsupernata ratiowasdeter each reactiona gradients.
thenumbero
the intact vir
ecules, arbitrarily expressed per 2,000N
mole-cules,weredetermined from eachgelasdetailed
inMaterials and Methods. Wherethetemplate
0 was not
saturated,
the number of L and NS}rl/N molecules rebound per 2,000 N molecules was
0 less than the number of L and NSproteinsfound
o0 inthe intact virion (Table 1).
However,inthecasewhere the templatewas
-v--NS/N saturated, there was excellent agreement
be-,,+,,A
aNS/N
tween the numbers of L and NS moleculesbound in vitro and in vivo (Table 1). In this
experimentLandNS proteinswerereboundto
purified template and the concentration of L
20 40 60 80 and NS proteins was shown to be saturating.
The
amount
of L andNSproteins
reboundperINITIAL RATIO35S/3H 2,000 N molecules in this experiment
ap-proachedclosely thenumber determined forthe
of LandNSproteinstotemplate intactvirion. These resultssuggestthatthe
re-Fingassayswassetupandprocessed binding is specific inthat saturationthe
Fig. and 2. Each tube contained 0.2
actionmixture,80Alofthe
XS-solubi-
binding
of L and NSproteins mimicked
the?ins, anddecreasingvolumes(80to10 bindingobservedintheintact virion.
ed template. Sufficient 0.01 M Tris- NSboundtotemplate in the absenceofL
pH 7.4, was added to each tube to protein. It wasofinterest to examine the sep-volumeto0.4ml.After separationof arate binding capabilitiesof the Land NS
pro-found'S-labeledproteinsonglycerol teins. The NS proteincanbe purifiedfrom the
pooledgradientfractions containing mixture of solubilized viral proteinsbyseveral and NSproteins were analyzed on cycles of column chromatography(seeMaterials
cmidegels. Thecountsof'Sin the L andMethods). A series of bindingassayswasset
werenormalizedtothecountsof 3H- upwithincreasingconcentrations ofNSprotein
'ein on each gel. These values were
the initial ratio of
5S-labeled
solubi- and constant amounts oftemplate.
AnSDS-ntproteins/3H-labeted
template.Ths
i polyacrylamide gel ofthe purified NS fraction ,minedby countingsmallsamples of showed 95% of the fraction to consist of NSiixturebefore layeringontheglycerol protein. Purified NSproteinbound to the
tem-plate in the absence of L protein (Fig. 4). Figure
scontained in 5 shows that the binding reaction was linear
rfL andNS proteins with increased protein concentration and that
ion If the number of L and NS NSprotein saturated the template.
proteins rebound to template
in
vitrogreatly
exceeded the number of L and NS proteins
normally carried by the virion, a nonspecific
aggregation might be suspected. On the other
hand,ifundersaturatingconditions the numbers
of L and NS proteinsreboundto thetemplate
wereapproximately equivalentto the virion
val-ues,specific rebindingwould besupported.
Forthese experimentsa3H-labeledaminoacid
mixturewasusedtouniformlylabelallthe viral
proteins.Asampleof viruswasdissociated with
the 2x HSS and separated by centrifugation
intoa supematant and pellet fraction.The sol-ubilized supematant fraction and either 0.1 or
0.05 ml of the pellet fraction which contained
thetemplatewereimmediatelyrecombined
un-der standard binding assay conditions and
ap-pliedtoaglycerol gradient.Thetemplate-bound
proteinswere thenanalyzedon
SDS-polyacryl-amidegels. Undissociatedvirus from the same
preparationwasalsoanalyzedonan
SDS-poly-acrylamide gel.Thenumbers of L and NS
mol-TABLE 1. NumbersofLand NSmoleculesbound
per2,000Nmoleculesa
Rebound Land NSproteins Pro- Mol Intact Subsaturationc
teins virionsh
Satura-0.1Tem-0.05Tem- tiond
plate plate
L 190,000 131 47 76 142
NS 40,000 267 160 224 230
aSee thetextforcalculations.
bIntact virions were solubilized and analyzed di-rectlyonpolyacrylamide gels.
'Samepreparationsasintact virion. The viruswas
solubilized,andsupernatantandpelletfractionswere obtained by centrifugation. Portions (80
pd)
of thesupernatant protein and0.1 or 0.05 ml of thepellet
fractionwereimmediatelyrecombined in the standard
bindingassay.
dPurified template and solubilized proteins
pre-paredfrom other virussampleswererecombined un-dersaturatingconditions inastandardbindingassay.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.501.59.245.59.248.2] [image:5.501.263.456.484.590.2]201
'Vo
c
E
0
OU 10
N
NS
- 10 20 30 40 50 60+
GEL FRACTION
FIG. 4. SDS-polyacrylamidegel electrophoresisof
template-bound NS protein from a binding assay
containing saturatingamountsof NS protein.
Bind-ingassay conditionsaredescribedin the legendto
Fig.5. 0.3
z
z
o 0.2 -z
0 z
0.1
_
0
O 0o
0 z
5 10
NS PROTEIN ADDED (cpmx10o3)
FIG. 5. Binding of NS proteintothenucleocapsid template.NSproteinlabeled with3Swaspurified by
chromatographyasdescribed in thetextanddialyzed against glycerolcolumn wash with 0.1 M NaCl. Tem-platelabeled with35Swaspreparedasdescribed in
thetext.Serial dilutionsofthe NSproteinweremade in 0.1 MNaCIcolumn wash. A 0.1-mlamountofNS
protein from each of thedilutions wasaddedto20 ,ul of template,60,1. oflxHSS,20!l1 of Tris,and 0.2 mlofprereactionmix(see text).Thebindingreactions
wereincubatedat31°C for30 min. Thetemplateand
boundproteins wereisolated onglycerol gradients
and analyzed on SDS-polyacrylamide gels. The countsin the NSproteinboundwerenormalizedto
thecounts intheNproteinon thegelsandplotted against the 35Scountsperminute in 20jlofeachof the dilutionsofpurifiedNS addedtotheassay.
Lprotein binding depended onthe
pres-enceof NSprotein.Incontrast tothe binding
capabilityofNSprotein, the Lprotein did not
appeartobindtotemplatein the absence of NS
protein. Forthisexperiment L-andNS-protein
fractions wereprepared by column
chromatog-raphy as described in Materials and Methods.
The L-protein fraction was free of NS protein
but contained substantial amounts of M protein. However, since the M protein did not rebind to
the template under these conditions,further
ma-nipulations to
purify
the extremely unstable Lprotein were not attempted. The NS fraction
usedwas atleast95% pure.
The L- and NS-protein fractions were
enzy-matically active when combined and tested in transcription assays. However, when tested alone each fraction was inactive, showing that the fractions were free of cross-contamination (datanotshown).
The L and NS fractions were added either
alone or in combination to purified template.
Table 2 shows that whentheL-proteinfraction
wasaddedalone,nobindingtothetemplatewas
seen.However, when Lproteinwasadded in the
presence of NS protein, Lprotein did bind to
the template. Thus, the binding of L protein seemedtobedependentonthe presenceof NS protein. Similaramounts of NSprotein bound to template in the presence or absence of L protein.
Requirements for
binding. Although
the bindingassaywasoriginally developed by using
conditionsoptimalfortranscription, further
ex-perimentsweredoneto seetowhatextentthese conditionswere
actually required.
Table 3com-paresbindingof L and NSproteins under
satu-ratingconditions in the presence and absence of
Mg2e
and nucleotides. The omission ofMg2e
andnucleotides did not
significantly
affect theamountof
binding
of the L andNSproteins
tothetemplate. Thus,
Mg2e
andnucleotideswerenotrequiredforbindingLand NS
proteins.
Another experiment was done to determine
whetherthebinding reaction wouldoccur atlow
temperature. After mixingthe components
un-dersaturating
conditions,
thebinding
reactionwasleft in the ice bucket insteadof
being
incu-bated at
310C.
Similar amounts of L and NS [image:6.501.55.215.59.218.2]proteins bound at4 and31°C
(Table
3).Thus,
TABLE 2. Binding ofLandNS proteins separately and incombination to purified template
Proteins bound(35S/3H)b
Fractions added'
L/N NS/N
L <0.005c
NS 0.232
L+NS 0.238 0.249
a35S-labeledL and NSfractionswereprepared and
recombined with3H-labeledpurified template as de-scribed in the text.
bTemplate-boundproteins isolatedonglycerol gra-dientswereanalyzedonSDS-polyacrylamide gels.
'NoLproteinwasdetected on thegel.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.501.75.213.272.445.2] [image:6.501.252.445.558.647.2]566
TABLE 3. RequirementsforthebindingofL and NSproteins
Proteins bound Incubation
(35S/3b
Reaction mixture temp0a H)(0C)
L/NNS/N
Completec 31 2.41 0.80
-Mg2e,
ATP, GTP, 31 2.22 0.78CTP
Complete 31 2.59 1.08
Complete 4 2.54 0.86
aAfter incubation at the indicated temperature,
all
gradientswereloaded inacold room andcentrifuged at40C.
bTemplate-bound proteins isolated onglycerol gra-dientswereanalyzedonSDS-polyacrylamide gels.
c35S-labeledsolubilizedviral proteins were prepared
asdescribed in the textand recombinedunder stan-dard assay conditions. All glycerol gradients corre-spond toprereactionmixtures withrespect to nucleo-tides andMg2+.
unlike transcription, the binding reaction
oc-curred at40C.
DISCUSSION
Thereconstitutionoftranscriptase activity by
recombination of template and soluble L and
NSproteins impliesthat theseproteinsrebind totemplatein afunctionally significantmanner. Therefore, the assay to detect this binding is
baseddirectlyontheconditions for
reconstitu-tion of transcriptase activity in vitro (7). The
greatest difficulty in developing an assay for
rebinding is the denaturation and aggregation of
the reaction components, especiallythe L
pro-tein. The assayreported here circumvents this
problem by isolating the nucleocapsid with
bound proteins on glycerol gradients through
whichaggregated materialspellet. Theamount
of bound L and NS proteins detected by the
assay saturates with approximately the same
number of L and NS molecules per 2,000 N
molecules as are present in the intact virion,
indicating that the binding detected is a specific
rebinding, similar tothat whichoccursin vivo.
Inaddition, theseresults suggest that the sites
onthetemplate available for binding L and NS
proteinsareprobablyfully occupiedinthe
pack-agedvirion.
In determining the numbers of L and NS
molecules bound in the intact virion, it was
alwaysnoted that the numbers ofNS molecules
weresignificantlygreater than the numbersof L
molecules. Of course, such calculations depend
on thestillunsatisfactory molecular weight
as-signments for L and NS proteins. The values
listed in Table 1 (131 molecules of L and 267
molecules of NS per 2,000 N molecules) were
calculated by using 190,000 and 40,000 as the
J. VIROL.
molecular weights for L and NS proteins and
result in an NS:L stoichiometric ratio of 2:1.
This ratiocanbecomparedtothe NS:L ratio of
1:1 reported for intact virions (and the in vitro
purified polymerase) by Naito and Ishihama
(11). These authors also used VS New Jersey
virusand molecular weights of 40,000 to 45,000
for NS protein and 190,000 for L protein.
Al-thoughreliable stoichiometric ratios await
bet-termolecular-weight assignments,especially for
NS protein, our data suggest some caution is
necessary in accepting the 1:1 stoichiometric
ratio forNS to L protein in the intact virion.
When NS and L proteins aresolubilizedfrom
thevirus, it isimpossibletodetermine to what
extenttheyexist insolutionaseitherindividual
proteins or asL:NScomplexes. It is clear that
when using the solubilized viral proteins as a
sourceof L andNS proteins, both L and NS are
bound to template, but analyzing the binding
from such acomplex,and as yetuncharacterized,
mixtureisdifficult. Our experimentsadding NS
and Lproteins separatelytothetemplatearea
start insortingoutthebindingcapabilitiesof L
andNS proteins. The results with the purified
NSprotein show that NS proteinbinds to the
template directly in the absence of L protein,
i.e., thetemplatemusthaveanaccessible
bind-ing site forNS protein.However, thebindingof
Lproteinappearstobedependentonthe
pres-enceof NSprotein.Since the inherentinstability
of Lproteinisaccentuatedby the manipulations
necessary to separate Lproteinfrom NSprotein,
this observation maysimply reflectthe
stabili-zation of L protein by NS protein. However,
since Land NSproteinscanbindtoeach other
(11), itispossiblethat Lproteinmaybind
indi-rectlytotemplatevia NSprotein.Alternatively, the NS protein may modify the template in
some way (e.g., exposing sites on the RNA)
whichallowsLproteintobinddirectly to
tem-plate.
Ourdataonthe numbers of bound L andNS
molecules have been arbitrarily calculated per
2,000 Nmolecules,notper virion.However,since
reported values for the numbers of N protein
per virion range from 1,000to2,000 (6, 12), we
would roughly estimate 65 or 130 L molecules
and134 or 267 NS molecules per virion. These
numbersmay becomparedtotheprobable
num-ber of promoter sites pergenome.Asingle
pro-moter site per genome is consistent with the
sequential transcription of VS virus genes
de-duced from UV-inactivation studies(4, 2,3),but,
atmost, the VS virus genome couldreasonably
beexpectedto have fiveor sixpromotersites,
onefor eachgene. Since wedetectmanymore
Land NS molecules
packaged
per virion thancould be boundtotheone tosixpromotersites,
on November 10, 2019 by guest
http://jvi.asm.org/
it is likely that the nucleocapsid has another
class ofbindingsites for thepolymerase.These
may be"reserve" sites, whichallow the virion to
package an extra supply ofpolymerase
mole-cules.Experiments in which thetranscriptionof
solubilized whole viruscanbe stimulatedatleast
threefold by the addition of extrapurified
tem-plate (unpublished data, S.U.E.) confirm the
suggestion thatVS virus packages more
polym-erase than can be used.
The two classes ofbindingsitesmay bequite
different. The promoterbindingsitesare
prob-ably restrictedtospecificsequences of RNAat
one orafewsitesonthe genome. The reserve
binding sites may be determinedbymore
gen-eral interactions with RNA and/or N protein
and may be distributed allalongthe
nucleocap-sid. Since our reactionswere done under
tran-scriptase conditions, rebinding to both sites probably occurs, but the characteristics of the
reaction are dominatedbybindingtothemore
numerous reserve sites. Further experiments,
including the binding ofpolymerase to naked
genomicRNA,areneededtofullycharacterize
the two types ofbinding.
ACKNOWLEGMENTS
This resech wassupported by PublicHealth Service grants AI-11722 (S.U.E.) and AI-05292 (M.G.M.) from the NationalInstituteofAllergyand Infectious Diseases. S.U.E. is therecipient of Public Health Service Research Career DevelopmentAward AI-00013 from the National Institute of Allergyand Infectious Diseases.
LITERATURE CITED
1. Abraham, G.,and A. K.Banerjee.1976.Thenatureof the RNA products synthesized in vitro by subviral components of vesicular stomatiti vius. Virology 71:230-241.
2.Abraham, G., and A. K. Banerjee. 1976. Sequential transiptionof thegenes of vesicular stomatitis virus. Proc.Natl.Acad. Sci. U.S.A.73:1504-1508.
3. Ball,L A. 1977. Transcriptionalmapping of vesicular stomatitis virus in vivo.J.Virol. 21:411-414.
4. Ball, iA.,and C. N. White. 1976. Order oftransciption of genesofvesicular stomatitis virus. Proc.Natl. Acad. Sci. U.S.A.73:442-446.
5. Bishop, D. H.L,S. U. Emerson, and A. Flamand. 1974. Reconstitution of infectivity and transcriptase activity of homologous andheterologousviruses: vesic-ular stomatitis(Indianaserotype), Chandipura, vesicu-lar stomatitis (New Jersey serotype), and Cocal Viruses. J.Virol. 14:139-144.
6. Bishop, D.H. L, and P. Roy. 1972. Dissociation of vesicular stomatitisvirus and relation of the virion proteins to the viraltranscriptase.J.Virol.10:234-243. 7. Emerson, S. U., and R. R. Wagner. 1972. Dissociation and reconstitution of the transcriptase and template activities of vesicular stomatitis B and T virions.J. Virol. 10:297-309.
8.Emerson, S. U., andR. R.Wagner. 1973. Lprotein requirement for in vitro RNA synthesis by vesicular stomatitis virus. J.Virol. 12:1325-1335.
9.Emerson, S.U.,and Y.-H.Yu.1975.Both NS and L proteins arerequired for in vitro RNA synthesis by vesicular stomatitisvirus. J. Virol. 15:1348-1356. 10.Hunt,D.M.,S. U.Emerson,and R.R.Wagner.1976.
RNA-temperature-sensitivemutantsofvesicular sto-matitis virus:L-protein thermosensitivityaccounts for transcriptaserestriction of group I mutants. J. Virol. 18:596-603.
11. Naito,S.,and A.Ishihama.1976.Function and structure of RNApolymerasefromvesicularstomatitis virus. J. Biol. Chem.251:4307-4314.
12.Nakai, T.,and A. F.Howatson.1968.The fine structure of vesicular stomatitisvirus.Virology35:268-281. 13. Reichman4nM E.,W. KLSchnitzleln,D.H.L.Bishop,
R. A.Lazzarini, S. T. Beatrice, and R.R.Wagner. 1978.ClassificationoftheNewJerseyserotype of vesic-ular stomatitis virus into two subtypes. J. Virol. 25:446-449.
14.Wagner,R. R. 1975. Reproduction ofrhabdoviruses, p. 27-30.In H.Fraenkel-Conratand R.R. Wagner (ed.), Comprehensive virology, vol. 4. Plenum Publishing Corp.,New York.
on November 10, 2019 by guest
http://jvi.asm.org/