Copyright©1977 American SocietyforMicrobiology Printed in U.S.A.
Maturation of Viral Proteins in Cells Infected with
Temperature-Sensitive Mutants of Vesicular Stomatitis
Virus
DAVID M. KNIPE,' DAVID BALTIMORE,* AND HARVEY F. LODISH
Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139
Received forpublication 10 September 1976
Maturation ofviral proteins in cells infected with mutants of vesicular stoma-titis virus was studied by surface iodination and cell fractionation. The move-ment ofG, M, and N proteins to the virion bud appeared to be interdependent. Mutations thought to be in G protein prevented its migration to the cell surface, allowed neither M nor N protein to become membrane bound, and blocked
formationof viral particles. Mutant G protein appeared not to leave the endo-plasmic reticulum at the nonpermissive temperature, but this defect was
par-tially reversible. In cells infected with mutants that caused N protein to be degraded rapidly or prevented its assembly into nucleocapsids, M protein did not bind tomembranesand G proteinmatured to the cell surface, but never entered structures with the density of virions. Mutations causing M protein to be degraded prevented virion formation, and G protein behaved as in cells infected by mutants in N protein. These results are consistent with a model of virion
formationinvolving coalescence of soluble nucleocapsid and soluble M protein with G proteinalready in the plasma membrane.
The pathways of maturation of the major structural proteinsof vesicular stomatitis virus (VSV) have been characterized (5). The
glyco-protein (G) isboth insertedintothe membrane ofthe endoplasmicreticulum andpartially gly-cosylatedsorapidlythatintermediatesare not
evident. Thefinal stagesof glycosylationoccur
afterthe Gprotein has migratedto
light-den-sity membranes; soon thereafter it appears on
the surfaceof the cell. Atearly times afterits
synthesis, the M protein is soluble; from this state itisprogressivelyincorporatedinto
mem-branous structures with the density of virions and thenquicklyappears inextracellular
viri-ons. Thenucleocapsid (N) protein is also
solu-bleafter its synthesisand is laterincorporated
intonucleocapsids that attach to the membrane prior tobudding into extracellular virions.
Temperature-sensitive mutants of VSV have
beenisolatedbyseverallaboratories (1, 2, 4, 10, 11), andmutants in some of the
complementa-tionclasses have been shown tobedefectivein certainmajor structural proteins (7-9). To char-acterize the effects ofthese mutations on mor-phogenesis of virions, we have examined the virus-specific structures that accumulate in cellsinfectedwith thesetemperature-sensitive mutants atthenonpermissive temperature.
1Present address: CommitteeonVirology,Universityof Chicago, Chicago,IL 60637.
MATERIALS AND METHODS
The origin and growth of the virus strains, cell fractionation, lactoperoxidase-catalyzed iodination ofthe cells, and othermethods used in thispaper have been described previously(5-7).
RESULTS
MigrationoftheGprotein tothe surface of
cells infectedwithtemperature-sensitive mu-tants.Itwaspreviouslyshown that the G pro-teinoftsM501(V) didnotappeartoundergothe final step(s) ofglycosylation because the
elec-trophoretic mobility of the proteindid not de-creaseduringachaseperiod (7).This
change
inmobilityhasbeen showntoinvolve the addition of sialic acid residues to the molecule several
minutespriortoitsappearanceonthe cell sur-face (6). We therefore tested whether the pro-tein encoded bythe mutant virus migrated to
thesurfaceof infectedcells. Thiswastestedin twoways,i.e.,surfaceiodination with
lactoper-oxidase and protease treatmentof [35S]methio-nine-labeled cells.
(i) iodination.The surfaceproteins of mock-infectedChinese hamster ovary cells labeled by lactoperoxidase-catalyzed iodination showed a pattern similartothat observedpreviously,
ex-cept that in this case some labeled protein,
migrating with bovine serum albumin and slightly slower than the VSV G marker, was 1149
on November 10, 2019 by guest
http://jvi.asm.org/
1150 LODISH
observed(Fig. 1). Thiswaspresumablythe re-sultof the serumproteinstickingtothe cells. In the cells infected with wild-type virus, a new
iodinated protein comigrating with virion G wasobserved onthe surface of the cells under allinfectionconditions (seethelegendtoFig.1
for an explanation of infectionconditions),and
thegreatest amount waspresent oncells kept
continuously at390C.
Iodination of cells infected with tsM301(III)
ateither 39 or 31'Crevealedthe presence of G protein on the surfaceofthe cells, but with a
greater amount at 390C than at 31'C (Fig. 1). These results indicatedthatincellsinfectedat
the nonpermissive temperature with a virus having an apparent mutationintheMprotein, the G protein maturednormallytothesurface
of the cells. It isofinterest to note that the G protein on the surface of cells infected with
rno
tsM301(III) at either 39 or 31'C did not comi-grate with the wild-type G protein, but mi-grated more slowly. This was previously ob-served with [35S]methionine-labeled proteins and isprobably due to differencesbetween our wild-type VSV and the parent of mutant tsM301(III) (7).
Themutantts045, agroupVmutantwhich, like tsM501(V), encodes aGproteinwhich
re-mains underglycosylated at 390C (data not
shown),waspreviously showntobedefectivein maturation of the G protein fromdense
mem-branestolight membranes (8). Weobservedno
Gprotein on thesurface of cells infected at 390C with ts045(V), but cells infected at 31'C
showeda considerable amount (Fig. 1). More-over,when cellsinfectedat390Cwereshiftedto
310C for only 1 h, a significant amount of G proteinaccumulated on the surface of thecells,
wt
M3O0(m)
045.-3
30
9 'S3
,
,mretne
.f
M 5)
1... ... !.)
:rt ..
rne,
t
)r
9m
Jne
FIG. 1. Surfaceiodination ofcellsinfected with temperature-sensitivemutants.Cells wereinfected with the indicated virus strain at a multiplicity of 10. At the termination of the infection, the cells were washed and iodinated as described in the text. The total cellular proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Protocol for infections:39°C,entireinfectionat39C-cells wereharvested
at5 hpostinfection; 31°C,entireinfectionat31°C- cells were harvested at 5 h postinfection; 39°C-- 31°C,
infectionat39°Cfor the first4h ofinfection and then shifted to 31°C for 1 h of incubation; 39°C-) 31 °C +
emetine, infectionat39°C for the first4 hof infection and then shifted to 31°C for 1 h of incubation in the presenceof 20 pg ofemetine perml (exposure time, 48 h).
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.501.71.452.275.578.2]showing that the defect was partially
reversi-ble. The appearance of G on the surface also
occurredinthepresenceof 20
gg
of emetine perml, aconcentrationsufficient to abolish all vi-rus protein synthesis. Hence, some of the G protein previously synthesized at 390C, but blocked in maturation within the cell, could move to thesurface of the cell once the temper-ature was lowered.
In addition, no labeled G protein could be found on the surface of cells infected with tsM501(V) at 390C, whereas at 31'C there wasa
detectable amount of G protein on the surface (Fig. 1). A small amount of G protein could be detected on the surface of cells within 1 h after cells, growing at 390C, were shifted to 31'C. This appearancealso occurred in the absence of protein synthesis, indicating that the defect waspartiallyreversible. Theincreasedlabel in the bands of the sample with emetine was
prob-ably dueto adarkerbackgroundbecause itwas
not apparent in other experiments. Thus, we
conclude that these two group V mutants are
defectivein maturation of the Gproteintothe surface of infected cells at the nonpermissive
temperature.
(ii) Protease treatment. To corroborate the
findings with lactoperoxidase labeling, we treated intact, infected cells with chymotrypsin toassay for the presence of the Gprotein on the cell surface. Infected cells were labeled with
[3:S]methioninefor 15 min and then incubated for an additional 60 min after the addition of excessunlabeled methionine. The infected cells werethen treated with chymotrypsin and pre-pared for gel electrophoresis. After a 60-min chase period, the G protein labeled at 39 or 31'C in cells infected with wild-type VSV was largely sensitive to prQtease treatment and thus was on the surface of the cells (Fig. 2).
The G proteinlabeled at 390C in cellsinfected
with tsM501(V) was not susceptible to protease treatment and thus not on the surface of the cells. Asevidencedby its protease sensitivity, it waspresent on the surface of the cells at 31'C. Thisconfirmed the conclusion from the
iodina-tsM
501(V)
310
39
31
tsM
301(I)
39
31
tsM 601
()39
31
- _
_-_ _ _ _
+ - + - + + - + - +
Chymotrypsin
treatment
FIG. 2. Protease treatment of intact cells infected with temperature-sensitive mutants of VSV. Cultures
wereinfected with theindicated virus strainatamultiplicityof10 and incubatedat31°C for5 h. At that time thecellswereresuspendedincomplete medium lackingmethionine,andone-halfofthemwereplacedat39°C and theremainderwereplacedat31'C.Aftera10-minwarming period,thecultureswerelabeledfor15min with[35S]methionine. The cultures were then incubated withexcess unlabeled methioninefor60min. The cellswerewashedand treated withchymotrypsin(1 mgofchymotrypsinpermlfor10minat37°C, except for
tsM301(III), whichwastreated with 10mgof chymotrypsinperml). Exposuretimes: wt, 24h;tsM501(V), 72 h; tsM301(III), 24h; tsM601(VI),24 h.
wt VSV
39
0G2N-NI
-Ns-
~~ - - 3-
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.501.50.444.316.579.2]tion results that the G protein encoded by tsM501(V) didnot mature properlyto the
sur-face of the cellsat390C.
Thistechnique was also applied to cells
in-fectedwith temperature-sensitivemutantsthat
weredefective in RNA synthesisatthe
nonper-missive temperature. RNA synthesis was
al-lowedto proceed at31'C, and the culture was
then shifted to 390C prior to labeling with
[35S]methionine. Thus,all of the labeled G
pro-tein would be synthesized and mature at the nonpermissive temperature. In contrast,
sur-face iodination would not distinguish newly synthesized G proteinfromGproteinputonthe
surface priortothe temperature shift. In cells infected with tsM601(VI) at 39°C, alarge
per-centage of the G protein wasremovedby
pro-teasetreatmentand thuswas onthesurfaceof
the cells. Because replication of viral RNA is low in these cellsat39°Cand alsothe Nprotein isdegradedrapidly(7),therecould beat mosta
small pool of nucleocapsids in these cells at 39°C. Therefore, it appears probable that
nu-cleocapsids have little role in maturation of the Gproteintothe surface of cells.
When cells infected with tsM301(III)at39or
31°Cwereexposedto1mgof chymotrypsinper
ml for 10 min, only 10 to 20% of the
[35S]-methionine-labeledG protein or iodinatedcell
surface G protein was removed (data not shown). Treatment of cells with 10mgof
chy-motrypsin perml for the sametime, however,
did remove [35S]methionine-labeled G protein
(Fig. 2) aswell asthe iodinated cell surface G
40
30
2 0
;.0
M protein N protein G protein
wild typeVSV 39,
30' label 30' chose
Ls
Hn
n-o I234 56 S
Gradient U
ructions P
a
,ln ,f1
23456SV
U P R
U
S
23456 S V
U P R
U S
protein (data not shown). Removal of the G protein from cells infected by the Glasgow strains ofVSV also required 10 mgofenzyme
per ml, suggesting that the parent of
tsM301(III)is the Glasgow strain and
support-ing the previousconclusion that tsM301(III) is the same mutant as tsG33(III). Using the
higher enzyme concentration, it is apparent
thatamutation of theMproteingenedoes not affect movement of the G protein to the cell
surface.
Fractionation of cellsinfected with
temper-ature-sensitive mutants. To examine the
ef-fectsof various mutationsonthe maturationof
the nonmutant proteins, we utilized the
frac-tionation procedure described previously (5). Cellswereinfectedwithwild-type and
temper-ature-sensitivemutantstrainsofvirus, labeled with[35S]methionine for30minat5h postinfec-tion, and subjected to chase conditions for 30 min at the permissive or nonpermissive tem-perature.
The proteins from each subcellular fraction
wererecoveredandsubjectedtosodiumdodecyl sulfate-polyacrylamide gel electrophoresis. The amountof eachviral proteinineachsubcellular fraction was expressed as apercentage ofthe
total viral proteinsinthe culture (Fig.3, 4,and 5).
(i) Cells infected with wild-type VSV. The
distribution of viral proteins in cultures in-fected with wild-type virus at 39 or 31'C was very similar to that described previously (5)
(Fig. 3). TheMprotein wasfound in the
cyto-M protein N protein G protein
4')
tsM30l(z) 39°C
30_ 30' lobel
30'chose
20 2
'O,0 .. _LE,,
,,..--23456 S V 23456 S V
Gradient U U
fractions P U P UR
S S
40 40
wildtypeVSV tsM301(z)
z ~~~~~~~310 311C
30-30'label 30 30'lobel
30'chose 30'Chose
20--
20-0 _rF~,
Ln
z~oH
Xn
123456 S V 123456 S V 123456 S V 123456 S V 123456 S V 23456 S V
FIG. 3. Fractionation of cells infected with temperature-sensitive mutants of VSV: wild-type and tsM301 (III)proteindistributions. Cultureswereinfectedwith the indicated virus strainatamultiplicity of10
at39or31'C. At5 hpostinfection the cellswereresuspendedin complete medium lacking methionine and
labeledfor 30 min. Excessunlabeled methioninewasadded and incubationwascontinuedfor 30min. The
cellswerethen fractionatedby the procedure described byKnipeetal. (5), with theuseof0.1M NaCl inthe initial centrifugationtoeliminate Maggregation. Theproteinswererecovered fromeach subcellular fraction
andsubjectedtosodium dodecylsulfate-polyacrylamide gel electrophoresis. Theamountof each protein in eachfractionwasdeterminedand expressedas apercentageof the totalamountof viral proteins in the culture.
_, .. _: "
I
20
v
R U S
123456 S V
u
P R u
s
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.501.117.404.424.580.2]M protein N protein G protein Mprotein N protein G protein
FIG. 4. Fractionation of cells infected with temperature-sensitive mutants of VSV: tsM501(V) and
tsM601(VI). Theseexperimentswereperformedasdescribedinthe legendtoFig.2,exceptthat cells infected
with tsM501(V) andtsM601(VI)wereused. The culture infected withtsM601(VI), whichwaslabeledat390C,
wasincubatedat31°Cfor thefirst5h of infection and shiftedto390C 10minpriortolabeling.
30 201
Z3 4 55 S V Z 5 4 5 b 5 V Z345 6 S V 2 345 6 5 V 2345 6 S V 2 3 4 5 6 S V
FIG. 5. Fractionation of cells infected with temperature-sensitive mutants of VSV: tsG41(IV) and tsM502(V). These experimentswereperformedasdescribedinthe legendtoFig.2,exceptthat cells infected withtsG41(IV)andtsM502(V)wereused. The cultures labeledat390Cwereincubatedat31'C for the first3h of infection, and one-halfwere shifted to 390C for 2 h of incubation prior to labeling atthe appropriate temperatures.
plasmicsupernatantand inasymmetrical
dis-tribution about fraction 4of the isopycnic
gra-dient. The membranousstructures infractions 3 and 4that contain Mproteinarebelieved to
benewlybudded virus attachedtothe cellsand buddingintermediates. Theonlydifference be-tweencultures infectedat39 and31'Cwasthe
observation that the percentage of soluble M protein wastwice as great at31'C asat390C, possibly the result of the increased degradation rateof theMproteinat390C (7). The Nprotein
wasfoundlargelyinnucleocapsidsinfraction 1
atboth temperatures.Nproteinwasalso found inthe cytoplasmicsupernatantandinasecond
peak of N protein-containing structures
cen-tered around fraction 4 in the gradient
mem-brane fractions. After thislongchaseperiod the
G protein is found mainly in the subcellular
fractions enriched inplasma membranes, i.e.,
fractions 5 and 6. There was alsosome G
pro-tein in fraction 4, presumably in virions at-tachedtocells.
(ii) Mutations in the M protein. The distri-bution of viral proteins in cells infected with tsM301(III) at390C showed that the Mprotein
wasfoundinsimilar fractionson theisopycnic
gradient as in wild-type virus-infected cells,
withapeakcentered around fraction4 (Fig. 3).
However,therewasverylittle Mproteininthe
cytoplasmicsupernatant, whichmayhave been
aresult of therapid degradationrateof the M protein at 390C (7). The N proteinwas found
almostentirelyin freenucleocapsids,and much less Nprotein floatedupintogradientfraction
tsG 4(IY)
30 31' - 39-C
I prior to
30'label
27 30' chose
0
~H
171 n--,n
-fh
2345 6 5 V 23 456 5 V 2 3456 S VI
toM502(Y)
so0_ 31,-_39-C _
30' label
20 ~~jchose30' j
° 2 3 4 5 6 5 V 23 4 5 6 5 V 2 3 4 56 S V
tsG41(I()2)
31' J 30' label at
30' chase
nHn
h
n Jrnits M502 (7)
31-30'label
30'chase
rflte ..n11 H 11 rI..
401~ 12 401
--0._ A I**1*-*III**
I
2
7
I . . I.' .. - - . I. .. . 11
G,.d,;,, u u . ; z G'ad'" u u 4 u 4 R
p p p ? -; act.... p p p
s s s w
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.501.100.392.69.234.2] [image:5.501.99.389.289.432.2]1154 BALTIMORE, LODISH
4ascomparedwith cellsinfected with wild-type virus at 390C or tsM301(III) at 31'C. The G protein was found largelyin fractions 5 and 6, apparently in the plasma membrane, because ofthe previous experiments (5) demonstrating the sensitivity of the G protein to protease treatment of intact cells. In addition, there werevirtually no viral proteinsfoundin
extra-cellular virus particlesproduced by these cells. Thus, thismutation prevented the formation of physical virus particles as well as infectious virus. This was consistent with the observa-tions of Unger and Reichmann (13), which showed that cells infected with tsG31, another group III mutant, incorporated labeled RNA intonucleocapsids, but not into virus particles. In cells infected withtsM301(III) at 31'C the distributions of the viral proteins were very similartothose incellsinfected withwild-type
virus. Much more Mprotein wasfoundin the cytoplasmic supernatant than at 390C, and much more Nproteinfloated up with the mem-branes inthe isopycnic gradient.
(iii) Mutationsin theglycoprotein. In cells infected with tsM501(V) at 390C, we observed that the largest amount of G protein was in
fractions 1 and 2, the fractions shown to be richestinendoplasmic reticulum (Fig. 4).Thus, the G protein may be defective in movement
from the endoplasmic reticulum. Consistent with this observation are the findings that at
390C only the G1 form of the glycoproteinwas
found and that noneof the G proteinwasfound
onthe cell surface (Fig. 1and 2). The N protein
was found mainly in fractions 1 and 2as free nucleocapsids. TheMprotein wasfound largely
in the cytoplasmic supernatant, with smaller amounts scatteredacrossthe densitygradient.
The amount of M protein in the supernatant has varied from 55 to 67% in several experi-ments, whereas only 20 to 30% of the wild-type protein was present in the cell supernatant. Therefore, the presence of G proteinonthe cell surface appears to have a role in the attach-ment oftheMprotein to membranes. At 31'C theGproteinmigratednormally to the plasma membrane fractions,andtheMand N proteins were found intheirnormal distribution about fraction 4. Total labeled protein accumulation
in extracellular virus was at least fivefold higher at 31'C, indicating a defect in particle maturation at thenonpermissive temperature. Although the data are not shown, similar
pro-tein distributions have been observed for tsO45(V), amutant having asimilar defect in maturationofthe Gprotein.
(iv) Mutations affecting the N protein.
Mu-tant tsM601(VI) was shown to encode an N
protein thatdegrades rapidly at 39°C, and the
mutantsynthesizes little 40S viral RNA, even at 31°C (7). Since this mutant is defective in RNA synthesis, we allowed RNA synthesis at 31°C for5handthen shifted thecultureto 39°C
prior to labeling with [35S]methionine. The amountofnucleocapsidsinthe cellswas there-fore determinedbythesize of thenucleocapsid poolat31°C.Asexpected,theamountoflabeled
Nprotein inthe cells labeled at39°C wasvery low due to its rapid degradation (Fig. 4). The G protein inthese cellswas foundlargely in the gradient fractions that contain plasma mem-brane (fractions5and6). However, the M pro-tein was largely in the cell cytoplasmic super-natant, indicating that it cannot stably attach
tomembranes.The percentage offree Mranged
from 67 to 75%, and the remainder ofthe M
protein was distributed across the gradient, withslightly larger amounts closer to the top of thegradient (fractions 5 and 6). At 31°C the N protein was partly in nucleocapsids, but, as
compared with wild-type VSV, an unusually large proportionwasin thecytoplasmic super-natant. This was not unexpected since the
amount of viral RNA replication was low at 31°C, but the N protein was stable. Thus, lessN
protein was bound to RNA inthese cells. The amount of soluble M washigh,presumably due
tothelowrateofvirus assembly because of the lackofnucleocapsids and the lower rate ofM
proteindegradation at31°C.
Althoughthe amount of viral proteins
assem-bled into virus from cells infected with tsM601(VI) and labeledat 39or 31°Cappears to be the sameasthatshowninFig. 4,inabsolute
amountsthe proteinsat31°Cwere two- to three-foldhigher dueto thedegradation of virtually all ofthe N proteinand significant amounts of
Mprotein at39°C.Thismadethepercentagesof
viral proteinsin extracellular particles at 39°C artificially high. We presume that cells infected with tsM601(VI) have a low level of intracellu-lar nucleocapsids at 31°C, which leads to the lowlevel of virusformation observed.
Incells infected with tsG41(IV) and labeled
at 39°C, a reduced amount of N protein was
presentinthe cells duetoitsrapiddegradation (Fig. 5). TheundegradedN proteinwas found
inlow amounts infraction 1,possiblyin nucleo-capsids, but much was in the cytoplasmic
su-pernatant. The Gprotein was largely in frac-tions 5 and 6 and thus presumably in the plasmamembranes. In contrast tocells infected
with tsM601(VI), a high percentage of the M protein and some G protein were in membra-nous structuresdistributedaboutfraction 4on
the isopycnic gradient, with normal levels in the cytoplasmic supernatant. At 31°C a
three-tofourfold-higher amountof viralproteinswas
on November 10, 2019 by guest
http://jvi.asm.org/
incorporated into extracellular virus, and the distributions of the viral proteins were very
similartothose of cells infected withour
wild-type virus.
We have included in this sectionexperiments describing the fractionation of cells infected withtsM502(V)because it is obvious from Fig.5
that tsM502(V) has a defect affecting the
as-sembly of N protein into nucleocapsids. In cells labeled at390C nearly all of the N proteinwas
presentinthe cytoplasmicsupernatantaswell
asmostof the viral M protein. Thus it appeared that the M protein could not stably bind to membranes in these cells. The G protein mi-grated normally to fractions 5 and 6, but seemedto represent alower percentageof the
total labeled viral proteins than normalatboth this temperature and 31'C. At 31'C a more
normal distribution of viral proteins was
ob-served, except that the amount of soluble N protein was still higher than wild type. This could mean that the defect is still partially
manifest even at the permissive temperature. Inspite of the fact thattsM502(V)wasassigned
to group V on genetic grounds, we have been
unable tofind anydefect in the maturation of
the G protein. The onlyapparentdefect is lower
levels of synthesis of G protein insome
experi-ments.
Temperature-shift experiments ofcultures infected with temperature-sensitive mutants. To explore whether the blockstovirion matura-tion defined above were reversible or not, we
incubated infected cells for 3 hat31°C, followed by2hat39°C, and the cellswerethenlabeled
for 30 min at 39°C with [35S]methionine. The cultureswerefurtherincubated withexcess
un-labeled methionine for 30 min. At that time one-halfof each infected culture wasremoved
and placedat0to4°C, and the remainderwas
shifted to 31°C for 1 h. At that time the cells
wereharvested from the cultures and their
cy-toplasm was fractionated into a supernatant
andapellet of particulate material,preparedas
in the previously described fractionation scheme. Theonly infected culture that incorpo-ratedasignificantamountof labeled viral
pro-teins into extracellular viral particles at39°C
was the culture infected with wild-type virus
(Table 1). After the shift intemperature to31°C the culture infected with wild-type virus
re-leased 2.5 times as much labeled protein in
extracellular virions. However, many of the cultures infected with temperature-sensitive
TABLE 1. Effectoftemperatureshiftonthe subcellular locationofproteins encodedbytemperature-sensitive
mutantsa
%of total:
M protein Nprotein Gprotein
Wildtype or mutant
Cellpel-
Cels-Cell
pel-Cels-Cell
pel- Cellsu-let perna- Virus let perna- Virus let perna- Virus
tant, tant tant,
wt, 390C 38 18 44 69 12.5 18 90 0 10
wt, 390C -*310C 15 7 78 42 12 46 45 45
tsM301(III), 390C 93 7 0 93 6 1 99.6 0 0.4
tsM301(III),390C- 95 0 5 76 13 10 88 0 12
310C
tsM501(V), 390C 29 71 0 81 19 0 100 0 0
tsM501(V), 390C - 54 42 4 90 6 4 100 0 0
310C
tsO45(V), 390C 49 51 0 19 28 3 100 0 0
tsO45(V), 390C - 64 11 24 75 16 9 89 0 11
310C
tsM601(VI), 390C 36 64 0 16 83 0 100 0 0
tsM601(VI), 390C- 36 41 22 15 85 0 97 0 3
310C
tsG41(IV), 39°C 69 26 6 49 51 0.7 94 3 3
tsG41(IV), 390C 51 12 37 83 15 2 90 0 10
310C
aCultureswereinfected with the indicated virusandincubatedat310Cfor3h. Atthattimethe cultures
wereshiftedto390Candfurtherincubated for2h. The cultureswerelabeledfor30minwith[35S]methionine and thenincubatedwithexcessunlabeledmethionine for 30 min. At that timeone-halfof the culturewas
removed andplacedat0to40C.Theremainderwastransferredto310C and incubationwascontinued for 60 min.The cellswerefractionatedintoacytoplasmicsupernatant anda100,000xgpelletof membranous and particulate material. Centrifugation wasperformedinthe presence of 0.1 M NaCl toeliminate Mprotein aggregation.
on November 10, 2019 by guest
http://jvi.asm.org/
mutant virus showed a much more dramatic increase intheamountof viral proteinsin
ex-tracellular virus,presumablydueto areversal of the temperature-sensitivedefect.
In cells infected with tsM301(III) the total
amount oflabeled viralproteins in virions in-creased approximately 20-fold after shifting to 31'C. Theamountof N andGproteinincreased
dramatically in extracellular virions after the shift-down intemperature, but very little of the
M protein that had not been degraded was
chased into virus, a result first observed by Lafay (8)fortsO89(III). Thus, Mprotein made
at 390C in cultures infected with tsM301(III)
was eitherin an aberrant structure or hadan
irreversibledefect. In a similarexperiment,the
extracellular virus titer increasedfrom5 x 104 PFU/mlinthe390C culture to8 x 106 PFU/ml
inthe infectionshifted to310Cfor 1h.Thus, it
appeared that infectious particles were
pro-duced after the shift intemperature, but little
M protein labeled at 390C was assembled into virus particles. This is evidence thatthe tem-perature-sensitive mutation of thetsM301(Ill) virus is adefect in theMprotein, asfirst noted by Lafay (8) forts089(III).
In cells infected with tsM501(V), the large
amountofsolubleMproteindecreasedafter the shift to 31'C, and a higher percentage of this protein wasboundtomembranes (Table 1). The G protein alsounderwent adecrease in mobil-ityas a resultofthe temperature shift, proba-bly a reflection of further glycosylation (not
shown). However, onlyasmall amountof viral protein assembled into extracellular virions
after thetemperatureshift,andthus thedefect
wasnotveryreversible.
The mutant tsO45(V), however, showed a more reversible phenotype as observed by
La-fay(8).After thetemperatureshift,the amount ofMbound to membranes increased, and the
amount of viral protein in virions showed an
eight- to tenfold increase. However, all of the labeled viral proteins were incorporated into virus in approximately the normal ratios,
in-cludingthe Gprotein,whichisblockedin mat-uration within the infected cell at 390C. This reversiblephenotype of theGprotein was also observed in the iodination experiments de-scribed above.
Incellsinfected withtsM601(VI) the
temper-atureshiftallowed anincrease ofincorporation of labeled proteins into virions. The small
amount of N protein in these cells was not incorporated into virus, but the M and G
pro-teins were. After the shift in temperature to 31°C wealso observed a decrease in the amount ofsoluble Min these cells.
The defect in virion maturation in cells in-fected with tsG41(IV)wasalso reversible. After the temperatureshift,alargeincrease in viral
protein assembly into extracellular virions oc-curred. ThelargeamountofMproteinbound to membranes in these cells decreased after the temperatureshift,andatleastpart of this ma-terialwasincorporatedintoextracellular virus.
Verylittle of theundegradedNprotein chased
outof the cellsduringthisperiod.
Thus, in many casesthe increase in soluble
M protein observed in the mutant virus-in-fected cells at the nonpermissive temperature
wasreversedbylowering the temperature, and this protein is presumably then incorporated into the membranes of the infected cells and intoextracellularparticles.
DISCUSSION
We have utilizedtemperature-sensitive mu-tantsofVSVin anattempttodefine the
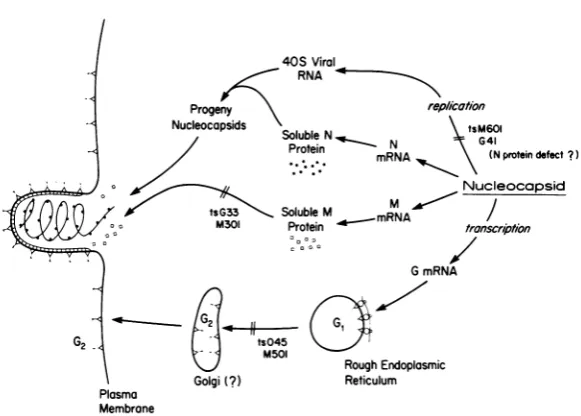
inter-relationships of the viral proteins in the proc-essesofvirionmorphogenesis. Certainaspects
of the separate pathways of maturation of the viralproteins shown schematicallyinFig.6are apparently independent of the other viral
pro-teins, suchasmigrationofthe Gproteintothe surface of the infected cell. Others, such as
binding of the M protein and nucleocapsid to
membranes, are the resultofinteractions
be-tweenviralproteins.
Effect of mutations in the glycoprotein.
Protease digestion and surface iodination ex-periments have demonstrated that the G pro-tein oftsM501(V)andts045(V) doesnot mature tothesurfaceofinfected cellsatthe nonpermis-sive temperature.Also, conversion of Gprotein
tothe sialylated
G,
form doesnotoccur under nonpermissive conditions. Cell fractionation studies have suggested that the G protein of tsM501(V) may be defective inmovementfrom the rough endoplasmic reticulum(fractions1 +2, Fig. 4). Thus, the lack of addition of the terminal sialic acid may be a result of the ina-bility of theproteintomigratetotheproper site
inthecell forglycosylationrather thanits
ina-bility toact as a receptor for thecarbohydrate residues. However, some mutant G protein does appear in fractions 5 + 6under nonpermis-siveconditions, andit is unclearwhether this reflects imprecision of thefractionation scheme
ortruemovement into smoothmembranes (see reference 5).
The defect of the G protein isinherently in-teresting because it implies that proteins once inserted into theendoplasmic reticulum are not passively transported bymembrane movement
tothe surface of thecell.Instead, some
on November 10, 2019 by guest
http://jvi.asm.org/
40S Viral
RNA
Progeny rep
Nucleocapsids
Soluble N N
Protein mRNA
*R*A
/ication
tsM601 G41
(Nprotein defect?)
Nucleocapsid
Soluble M mRNA / Protein transcription
I _
G mRNA
RoughEndoplasmic
Reticulum
FIG. 6. Schematic diagram illustrating the pathways of maturation of the majorstructural proteins of VSVand theproposedsiteofblock in virionassemblyforcertain temperature-sensitive mutants.
tionbetween the G protein and cellular struc-tures, possiblytransport proteins, mustoccur.
Whateverthe defectmayactually be,aportion
ofthe G protein oftsO45(V) can move to the surface of the celloncethetemperatureis
low-ered, suggestingthattheproteincanfoldtoits proper conformation and then mature
nor-mally.
Effect ofdefectsinassembly of nucleocap-sids. Two mutantsthat encodealabile N
pro-tein andsynthesize little40Sviral RNAatthe
nonpermissive temperature yielded somewhat
different results in terms of the viral protein
structures that accumulate in cells infected
with these viruses at the nonpermissive tem-perature. The maturation of the G protein to plasma membranes appeared to be normal in
bothcases, and thusthere was noevidence for
anyrole of thenucleocapsids inmaturation of the Gprotein. However, incells infected with tsM601(VI) nearly all of the M protein was
soluble, whereas in cells infected with
tsG41(IV) alarge percentage of the M protein wasinmembrane-boundstructureswith
inter-mediate density. Thesemay have been actual
intermediates of budding because at least a
portionofthesestructurescouldbechased into virionsafterashift intemperature to31°C.The
differencebetween thesetwosituationsmaybe
explained by differences in the actual muta-tions of the viruses. In cells infected with tsM601(VI)thereareprobablyveryfew nucleo-capsids in the cells at any temperature, and
under these conditions the M protein maynot
be able to bind stably to membranes. On the other hand, tsG41(IV) accumulated large amounts of 40S RNA at 31°C, and while
allowing synthesis of mRNA at 310C we must
be allowing the accumulation of significant amountsofnucleocapsids. These maybe
capa-ble ofbinding to membranes with the M
pro-tein, but incapable of budding from the cell until thetemperatureis lowered.
Incells infected with tsM502(V)noneofthe N
protein is assembledintonucleocapsids. Again,
verylittle of the M protein is bound to
mem-branes, suggesting that the M proteincannot formastablecomplexontheplasmamembrane
with only the glycoprotein. This analysis is
complicated, however, bythe fact thatwe
can-notruleoutthepossibility ofasecondmutation
inthe G proteinsuggested byits assignmentto complementationgroupV.
Effect of mutationsinthe Mprotein.The M
protein incells infected withtsM301(III)atthe nonpermissive temperature is degraded at a
ratethree-tofourfold faster than theMprotein in cells infected with wild-type virus (7). The residual undegraded tsM301(III) M protein is almostexclusivelymembranebound,and much of it bands at the density of whole virions. These structures may not be normal budding
intermediates because none of the M protein
couldbe chased into virionsuponashift-down to the permissive temperature. Furthermore, protease treatment of intact cells couldnot re-movethe membrane-boundMproteins, andno
buds wereevident on the cellsby electron
mi-ts045
M501
Golgi(?)
1157
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.501.106.397.70.278.2]KNIPE,
croscopy (unpublished data). We are presently
uncertain as to the exact identity of these M
protein-containing structures. The
tempera-ture-sensitive lesion, apparently in the M
pro-tein, had no effect on the migration of the G
protein tothecell surface, and thus there was no evidence fora role ofthe M protein in the
maturationof the G protein.
ACKNOWLEDGMENTS
We gratefully acknowledgethe technical assistance of Martin Brock.
D.K. wassupported by a National Science Foundation predoctoral fellowship during partof this work and aPublic Health Service traineeship during the remainder. D.B. is anAmerican Cancer Society researchprofessor. H.F.L. was therecipient of Public Health Service research career devel-opment award GM-50175 from the National Institute of General Medical Sciences. This work was supported by Public HealthService grants AI-08814 and AI-08388 from theNational Institute of Allergy and Infectious Diseases, American Cancer Society grant E559, and Public Health Service grant CA-12174 from the National Cancer Institute.
LITERATURE CITED
1. Flamand, A. 1969. Etude des mutants thermosensibles du virus de la stomatite vesiculaire mise en point d'untest de complementation. C.R. Acad. Sci. Paris 268:2305-2308.
2. Flamand, A. 1970. Etude genetiques du virus de la stomatite vesiculaire: clasement de mutants thermo-sensibles spontanes en groupes de complementation. J.Gen.Virol. 8:187-195.
3. Garoff, H.,and K. Simons. 1974. Locationof thespike glycoproteins in the Semliki forest virus membrane. Proc. Natl. Acad. Sci. U.S.A. 71:3988-3992.
4. Holloway, A. F., P. K. Y. Wang, and D. V. Cormack. 1970. Isolation and characterization of temperature-sensitive mutants ofvesicular stomatitis virus. Virol-ogy 42:917-926.
5. Knipe, D. M., D. Baltimore, and H. F.Lodish. 1977. Separate pathways of maturation of the major struc-tural proteins ofvesicular stomatitis virus. J. Virol. 21:1128-1139.
6. Knipe, D. M., H. F. Lodish, and D. Baltimore. 1977. Localization of two cellular forms of the vesicular stomatitisviral glycoprotein. J.Virol.21:1121-1127. 7. Knipe, D., H. F. Lodish, and D. Baltimore. 1977.
Anal-ysis of the defects of temperature-sensitive mutants of vesicular stomatitis virus: intracellular degrada-tionofspecificviralproteins. J. Virol. 21:1140-1148. 8. Lafay, F. 1974.Envelope proteins of vesicular
stomati-tisvirus:effectof temperature-sensitive mutations in complementation groups III and V. J. Virol. 14:1220-1228.
9. Ngan, J. S. C., A. F. Holloway, and D. V. Cormack. 1974.Temperature-sensitive mutants ofvesicular sto-matitis virus:comparison of the in vitro polymerase defectsof group I and group IV mutants. J. Virol. 14:765-772.
10. Pringle, C. R. 1970. Genetic characteristics of condi-tional lethal mutants of vesicular stomatitis virus induced by 5-fluorouracil, 5-azacytidine, and ethyl methane sulfate. J. Virol. 5:559-567.
11. Pringle, C. R. 1970. Theinductionand genetic charac-teristics ofconditional lethal mutants ofvesicular stomatitisvirus, p. 567-582. In R. D.Barryand B. W. J. Mahy (ed.), The biology oflarge RNA viruses. Academic Press Inc., London.
12. Rettenmier, C., R.Dumont, andD. Baltimore. 1975. Screeningprocedureforcomplementation-dependent mutantsof vesicular stomatitis virus. J.Virol. 15:41-49.
13. Unger, J. T., and M. E. Reichmann. 1973. RNA synthe-sis intemperature-sensitive mutantsof vesicular sto-matitis virus. J. Virol. 12:570-578.
on November 10, 2019 by guest
http://jvi.asm.org/