Copyright © 2002, American Society for Microbiology. All Rights Reserved.
NOTES
Mutations That Confer Resistance to Template-Analog Inhibitors of
Human Immunodeficiency Virus (HIV) Type 1 Reverse Transcriptase
Lead to Severe Defects in HIV Replication
Timothy S. Fisher, Pheroze Joshi, and Vinayaka R. Prasad*
Department of Microbiology and Immunology, Albert Einstein College of Medicine, Bronx, New York 10461
Received 4 October 2001/Accepted 11 January 2002
We isolated two template analog reverse transcriptase (RT) inhibitor-resistant mutants of human immu-nodeficiency virus (HIV) type 1 RT by using the DNA aptamer, RT1t49. The mutations associated, N255D or N265D, displayed low-level resistance to RT1t49, while high-level resistance could be observed when both mutations were present (Dbl). Molecular clones of HIV that contained the mutations produced replication-defective virions. All three RT mutants displayed severe processivity defects. Thus, while biochemical resis-tance to the DNA aptamer RT1t49 can be generated in vitro via multiple mutations, the overlap between the aptamer- and template-primer-binding pockets favors mutations that also affect the RT-template-primer interaction. Therefore, viruses with such mutations are replication defective. Potent inhibition and a built-in mechanism to render aptamer-resistant viruses replication defective make this an attractive class of inhibitors.
Due to its central role in human immunodeficiency virus type 1 (HIV-1) viral replication, reverse transcriptase (RT) is a major target for antiviral chemotherapy. Most drug combina-tions used currently in highly active antiretroviral therapy (HAART) for AIDS include at least one anti-RT drug. While HAART can often suppress viral replication to undetectable levels, it does not prevent the emergence of drug-resistant variants (18). Despite the availability of two classes of anti-RT drugs, development of new, more potent, and less toxic an-ti-RT drugs remains a crucial goal. Among new agents that potently block HIV-1 replication are nucleic acid-based inhib-itors that target reverse transcription, such as tRNA decoys (27), antisense (10) and phosphorothioate nucleic acids that
bind tRNA3Lys (14), and aptamers that mimic RT’s nucleic
acid substrate (35).
High-affinity DNA and RNA ligands, or aptamers, were isolated via the systematic evolution of ligands by exponential enrichment (SELEX) procedure (36) for a number of HIV targets, including the integrase (1), nucleocapsid (9), Tat (37), Rev (17, 21), and RT proteins (31, 35). Aptamers targeting HIV-1 RT bind with high affinity and potently inhibit its RNA-dependent DNA polymerase (RDDP) activity (31, 35). This inhibition is selective to HIV-1 RT, with no effect on the activities of related RTs such as those of avian myeloblastoma virus RT and Moloney murine leukemia virus RT (31).
The X-ray crystal structure of HIV-1 RT complexed with an RNA aptamer shows that the aptamer-binding surface partially overlaps the binding surface of template-primer substrates (20). Furthermore, the inhibition of RDDP activity by such
aptamers was found to be competitive with respect to the template-primer (11, 12). Taken together, these results suggest that both DNA and RNA aptamers mimic the enzyme’s natu-ral nucleic acid substrate. Therefore, aptamers that target HIV-1 RT are referred to here as template-analog RT inhib-itors (TRTIs). The presence of a large surface on RT for binding TRTIs may require multiple mutations to generate resistance, reducing the likelihood that a TRTI-resistant vari-ant will emerge. Additionally, since the TRTI- and template-primer-binding surfaces overlap, mutations that confer TRTI resistance will likely target the template-primer-binding cleft and may incapacitate RT.
In order to determine the consequences of TRTI resistance to RT function and to HIV replication, we isolated two HIV-1 RT mutants displaying resistance to the DNA aptamer RT1t49 (31) via a phenotypic screen of a library of random mutations. Single mutations conferred low-level resistance, while multiple mutations were necessary for high-level resistance. Interest-ingly, both mutations that conferred resistance to RT1t49 lie close to a key functional element of HIV-1 RT, the minor groove binding track (MGBT) (8), and cause severe defects in RT polymerase processivity. Cell culture virus replication stud-ies showed that the mutations, singly or together, cripple the virus, precluding their emergence in vivo.
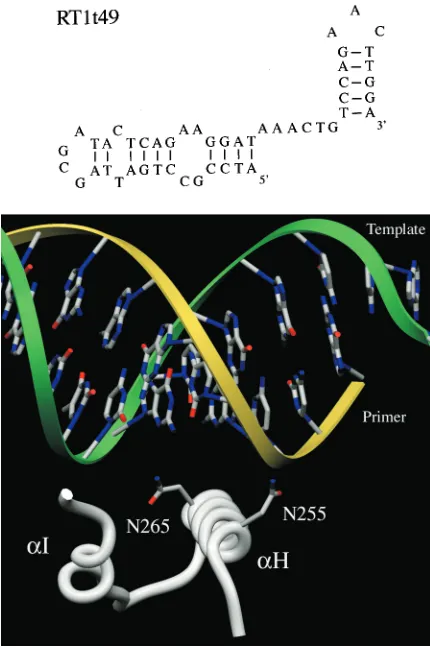
Isolation of TRTI-resistant RT mutants.We sought mutants of HIV-1 RT that were resistant to inhibition by the DNA aptamer RT1t49. The secondary structure of RT1t49, as pro-posed by Schneider et al. (31), is shown in Fig. 1, top. RT1t49
binds HIV-1 RT with high affinity (Kd⫽4 nM) and potently
inhibits its activity. A random library of mutations (at amino acid residues 1 to 312) generated by error-prone PCR (26) in a bacterial HIV-1 RT expression vector, pHRTRX2 (34), was screened via the in situ colony-screening assay as previously described (29, 30) for RDDP activity in the presence of 25 nM
* Corresponding author. Mailing address: Department of Microbi-ology and ImmunMicrobi-ology, Albert Einstein College of Medicine, 1300 Morris Park Ave., Room GB 401, Bronx, NY 10461. Phone: (718) 430-2517. Fax: (718) 430-8976. E-mail: [email protected].
4068
on November 8, 2019 by guest
http://jvi.asm.org/
RT1t49: a concentration that inhibits wild-type RT RDDP
activity to 肁95% in vitro. From among ⬃50,000 colonies
screened, we isolated two clones. Sequence analysis of the entire RT revealed that each clone carried a single base
change, AAT3GAT, leading to a substitution of aspartate (D)
for asparagine (N) at either codon 255 or 265 of HIV-1 RT.
Residues 255 and 265 are found within the␣H helix of the
thumb subdomain, which forms a track for the minor groove of the template-primer duplex during enzyme translocation (Fig.
1, bottom) (8, 13). The surface of␣H helix facing the dsDNA
minor groove makes numerous contacts with the template and primer two to six base pairs from the active site and has been shown to play an important functional role in HIV-1 RT tem-plate-primer-binding, translocation, and frameshift fidelity (6– 8).
Biochemical properties of TRTI-reistant mutant RTs. In order to evaluate the influence of N255D and N265D substi-tutions on RT function, we generated the purified het-erodimeric RTs containing each of the mutations separately or together in the p66 subunits, as described previously (23). The
enzymatic activities of the mutant RTs in steady-state nucleo-tide incorporation assays, using poly(rA)-oligo(dT)
template-primer, were found to be not severely affected (Vmax/Km.dTTP
values being 1.2, 0.8, 0.45, and 0.52 for the wild type [WT]and for the N255D, N265D, and Dbl mutants, respectively). We
also determined the Kmfor 16S rRNA annealed to a DNA
primer (VP200) and found that the mutant RTs’ abilities to interact with normal template-primer were minimally affected,
with theKm.T-Pvalues being 6.5 nM (WT), 17.2 nM (N255D),
7.7 nM (N265D), and 37.3 nM (Dbl), respectively. As these
data show, theKm.T-Pvalues are within sixfold of that of the
WT enzyme.
We determined the degree of resistance conferred by each of the two mutations, using 16S rRNA annealed to a DNA primer (corresponding to nucleotides 885 to 906 of 16S rRNA) as template-primer, as described previously (23). In reactions using increasing amounts of RT1t49, the N255D and N265D substitutions each conferred low-level resistance (50%
inhibi-tory concentration [IC50] of 7.9 nM and 17.4 nM compared to
1.6 nM displayed by WT RT) of about 5- and 11-fold increase, respectively, over that of WT RT. The N265D substitution consistently conferred a higher level of resistance to RT1t49 than N255D on all template-primers tested (data not shown). Interestingly, when placed together, the two mutations acted synergistically in conferring an approximately 150-fold
resis-tance compared to that of the WT enzyme (IC50of 245 nM).
Thus, single mutations conferred low-level resistance to the DNA TRTI RT1t49, while multiple mutations are needed for high-level resistance.
Resistance is due to reduced affinity to RT1t49.For a com-petitive inhibitor, resistance is mediated by reduced binding. To test possible alterations in binding affinities, we performed electrophoretic gel mobility shift analyses with purified WT and mutant RTs (Fig. 2). Increasing concentrations (0.1 nM to
2.0M) of WT and mutant RTs were incubated at 25°C for 10
min with 5⬘-32P-labeled RT1t49 in 50 mM Tris-Cl, pH 8.0, 25
mM KCl, 10% glycerol, and 1 mM dithiothreitol. When the reaction mixtures were electrophoresed on a nondenaturing polyacrylamide gel electrophoresis (PAGE) gel (8% polyacryl-amide [8% PAGE]), increasing the ratio of WT RT to that of
[image:2.587.52.267.72.396.2]FIG. 1. (Top) Proposed secondary structure of DNA TRTI RT1t49 (31) (reprinted with permission from the American Chemical Society). (Bottom) Location of the N255 and N265 residues near the template-primer. Ribbon diagram of helix H and I relative to template-primer, showing the location of residues in RT1t49 with resistance mutations in the␣H helix of the thumb subdomain. N255 and N265 are located near the MGBT (8). The illustration was created with the program SETOR (16) by using the X-ray crystallographic coordinates of a trapped catalytic complex of HIV-1 RT with the DNA template-primer and ddTTP (19).
FIG. 2. Electrophoretic mobility shift assays to determine RT1t49-binding affinities of WT and mutant RTs. Note that the concentrations of the mutant RTs ranged from 0 to 100 nM, while that of the WT enzyme ranged from 0 to 10 nM. The migration positions of both free and complexed DNA aptamer RT1t49 are indicated on the right of the panel. All reactions were resolved by nondenaturing 8% PAGE.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:2.587.328.513.73.202.2]the 5⬘-P32-labeled RT1t49, resulted in a shift of the free RT1t49 into a slower-migrating complex. In contrast, at the same protein concentrations, the N255D, N265D, and Dbl mutant RTs failed to form comparable amounts of this com-plex (Fig. 2). Phosphorimaging analysis of the gels helped us quantitate the shifted complexes and determine the
dissocia-tion constants (Kd) for each enzyme-RT1t49 complex. The WT
RT had the highest affinity (Kdof 0.8⫾0.05 nM). The N255D
and N265D mutant RTs displayed 57- and 115-fold higherKd
values than WT RT with RT1t49 (Kdvalues of 45.4⫾5.6 nM
and 91.8 ⫾ 3.9 nM, respectively). Taken together, the two
mutations led to a greater than 2,500-fold increase in theKd
compared to that for the WT enzyme (Kd⫽ ⬎2,000 nM). Even
at the highest concentration of enzyme tested (2.0 M), no
more than 20% of the DNA aptamer was found to be in complex with the Dbl mutant RT (T. S. Fisher and V. R. Prasad, unpublished results). The loss of affinity of mutant RTs to the aptamer DNA is not due to nonspecific loss of nucleic acid binding, as shown by the facts that the mutant RTs display
robust enzymatic activities and their Km values for normal
template-primers, presented above, were all within sixfold of each other. Thus, it appears that there is a direct correlation between TRTI binding and sensitivity to inhibition by RT1t49.
Effects of mutations on viral infectivity and replication ki-netics.Since the TRTI resistance mutations were shown to be located within the template-primer-binding tract, these muta-tions could affect RT function and, hence, viral replication. Therefore, we introduced N255D and N265D mutations singly or together (Dbl) into the full-length infectious molecular
clone, HIVR3B. To generate viral stocks, 293T cells (3⫻106)
were transfected with HIVR3B DNA by lipofection
(Gene-PORTER; Gene Therapy Systems, San Diego, Calif.). Forty-eight hours posttransfection, culture supernatants were tested
for p24Gagprotein production by using the HIV-1 p24
enzyme-linked immunoassay (ELISA) (NEN Life Science Products, Boston, Mass.). We assessed the effect of HIV-1 RT TRTI resistance mutations on viral replication in cell culture. Jurkat
T cells (106), maintained in RPMI 1640 medium, were infected
with equal amounts of virus (40 ng of p24 each), and the kinetics of viral growth were monitored for up to 21 days
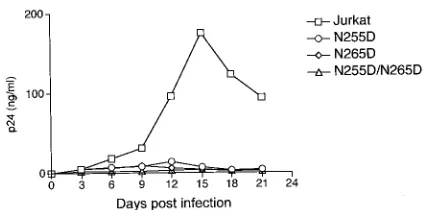
postinfection via ELISA. Viruses containing the TRTI resis-tance mutations grew much less efficiently than WT virus in T cells. Twenty-one days after infection, we observed that all three viruses showed no signs of viral replication (Fig. 3). These results indicate that mutations in HIV-1 RT that confer resistance to RT1t49 also cause a severe reduction in viral infectivity and replication in cell culture.
Biochemical defects resulting from TRTI resistance muta-tions. Since the TRTI-resistant RT mutants were isolated based on biochemical function, the molecular defect responsi-ble for a compromised replication of viruses containing the same mutations is not due to a gross defect in polymerase activity. Therefore, we wished to determine whether specific polymerase properties of RT were defective.
Since the␣H helix of HIV-1 RT is known to play an
impor-tant role in processive DNA synthesis (7), we asked whether the severe defect of viruses carrying TRTI resistance mutations was possibly due to processivity defects. We tested the proces-sivity of WT and mutant enzymes by using an M13
single-stranded DNA template annealed to 5⬘-P32-labeled sequencing
primer⫺47 (New England Biolabs, Beverly, Mass.). For each
set of reactions, equal inputs of the different enzymes (1 U each) were used. To detect single-cycle polymerization effi-ciency, an enzyme trap [poly(rA)-oligo(dT)] was included to prevent rebinding of dissociated RT molecules. Reaction mix-tures containing RT and 5 nM template-primer were preincu-bated for 5 min at 37°C, and polymerization was initiated by
the addition of 50M dinucleoside triphosphates (dNTPs) and
a 20-fold molar excess of poly(rA)-oligo(dT) trap (37°C for 15 min). Under these conditions, each band detected by denatur-ing PAGE should be the product of a sdenatur-ingle cycle of binddenatur-ing, elongation, and dissociation. The trap effectiveness was deter-mined by preincubating RT with trap and then starting the reactions by the addition of dNTPs and template-primer. Such reactions led to no primer extension (data not shown). In parallel assays that facilitated multiple rounds of synthesis, only WT RT produced products longer than 600 nucleotides (nt) (Fig. 4, lane 1). Although neither N255D RT nor N265D RT yielded DNA products comparable to those by WT, they both generated products longer than 300-nt under these con-ditions (Fig. 4, lanes 2 and 3). However, under concon-ditions of single-cycle polymerization, both single mutants had severe defects in processivity compared to WT RT (Fig. 4, lanes 5 to 7). In single-cycle reactions, WT RT synthesized products longer than 500 nt, in comparison to few products of 100 and 130 nt for N255D and N265D mutant RTs, respectively. In contrast, the Dbl mutant RT displayed little to no DNA-de-pendent DNA polymerase activity (Fig. 4, lane 8). Even when multiple rounds of polymerization were allowed, the Dbl en-zyme was predominantly blocked after extending the primer
only⬃150 nt (Fig. 4, lane 4). Altogether, the TRTI-resistant
RTs appeared to dramatically lower HIV-1 RT processivity. We have described data showing that mutations conferring in vitro resistance to the TRTI RT1t49 can be generated via an in situ phenotypic screen. The SELEX procedure favors aptamers that bind their target with the highest binding affinity. HIV-1 RT aptamers tend to display extraordinarily high
affin-ity (Kd,⬃25 pM) (22). It has been argued that resistance to
aptamers will be hard to achieve, since the target protein has multiple contact points for the ligand distributed over the
en-FIG. 3. Replication kinetics of viruses carrying TRTI resistance mutations. Jurkat T cells were infected on day 0 with a WT HIV-1 R3B virus or with viruses carrying RT mutations shown to confer resistance to TRTIs in vitro. Aliquots of culture supernatant were collected every day for 21 days to monitor viral replication, and equivalent amounts of RPMI 1640 were added to the cultures to replace the volumes re-moved. Viral production was monitored by measuring the concentra-tion of HIV-1 p24 antigen in the culture supernatant. Viral input was normalized according to the HIV-1 p24 antigen content of transfected 293T cell supernatants.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:3.587.56.268.71.176.2]tire binding pocket (35). Therefore, resistance to such com-pounds would likely require multiple mutations or would need to target residues that serve as pivotal contact sites between RT and its nucleic acid substrate. Consistent with this idea, mutations conferring resistance to the TRTI RT1t49 were
found within a crucial structural element of the template cleft, the MGBT (Fig. 1, bottom). In addition, our findings suggest that single mutations, in fact, are not able to confer significant levels of resistance in vitro.
When growth curves of mutant viruses were determined in T-lymphoid cells, all mutant viruses produced very low to un-detectable levels of p24 in the medium, indicating severe rep-lication defects (Fig. 3). Mutations within HIV-1 RT that con-fer high-level resistance to TRTIs (Dbl) result in the greatest defect in both the biochemical function of the resistant enzyme (overall activity as well as processivity) and in the replication competence of viruses carrying such mutations.
We have investigated the RT defects that likely result in severely compromised virus replication. Both N255 and N265
are within the␣H helix of the thumb subdomain thought to
form part of the MGBT (8), or translocation track (13), which facilitates processive RT movement along the template-primer duplex. The MGBT consists of residues Q258, I94, G262, W266, and Q269, and mutation at these residues is accompa-nied by a decrease in template-primer-binding affinity, proces-sivity, and frameshift fidelity (6–8). Similar to MGBT muta-tions, the TRTI resistance mutations isolated in this study resulted in a severe defect in processivity (Fig. 4). Reduced processivity (3, 32) due to the didanosine resistance mutation L74V or the lamivudine resistance mutation M184V has pre-viously been shown to affect HIV replication. In comparison, RTs resistant to the DNA TRTI RT1t49 displayed much lower processivity than either the didanosine-resistant L74V or lami-vudine-resistant M184I/V RTs (Fig. 4) (3, 32). Therefore, it is not surprising that viruses expressing RTs containing TRTI resistance mutations were severely defective for infectivity and replication.
Based on our results, we speculate that a key problem in developing anti-HIV agents, that of viral resistance, is less likely to be a problem in the case of TRTIs caused by pertur-bation of critical functions intrinsic to HIV-1 RT. It remains to be seen whether mutations conferring resistance to TRTIs targeting nonessential regions of the template-binding cleft can arise. Although our results are promising, in order for TRTIs to effectively function as anti-HIV inhibitors, both their in vivo stability and delivery must be addressed. In recent years, sev-eral strategies have been developed to increase the in vivo stability of DNA- and RNA-based inhibitors (2, 28). These techniques have resulted in several thousandfold increases in the stability of RNA and DNA aptamers in serum and cellular extracts. There has also been considerable interest in deliver-ing nucleic acid-derived drugs in vivo. For example, liposomes have been used effectively to deliver both traditional anti-HIV agents such as HIV protease inhibitors and nucleoside RT inhibitors, as well as nucleic acid-based inhibitors, including an anti-HIV ribozyme and an anti-Rev aptamer, into HIV-in-fected cells (15, 25). Alternatively, gene delivery approaches that are being developed for the expression of therapeutic nucleic acids within hematopoietic stem cells may lead to the development of HIV-1 RT-specific RNA aptamers as potent anti-HIV agents (4, 5, 24, 33). By combining both the potent inhibition of reverse transcription with a built-in mechanism for TRTI resistance, leading to a loss of viral fitness with these strategies, TRTIs have potential as anti-HIV inhibitors.
FIG. 4. Processivity of WT and TRTI-resistant HIV-1 RTs. Prod-ucts synthesized by enzymes in multiple rounds (⫺trap) or in a single processive cycle (⫹trap) are shown. Reaction mixtures contained the M13mp18 template annealed to the⫺47 primer, RT heterodimer, and dNTPs. The positions of size standards are indicated on the right. Reaction products were resolved by denaturing 5% PAGE.
on November 8, 2019 by guest
http://jvi.asm.org/
The research described in this report was supported by a Public Service grant to VRP (RO1 AI30861). T.S.F. acknowledges support from an institutional predoctoral training grant (NIH T32-GM07491). We thank Jurgen Brojatsch, William Drosopoulos, Scott Garforth, and Maria Dolores Iglesias for reading the manuscript, Matthew Ro-den for help in generating the Ribbons diagram, and William Droso-poulos for the randomized RT expression library.
REFERENCES
1.Allen, P., S. Worland, and L. Gold.1995. Isolation of high-affinity RNA ligands to HIV-1 integrase from a random pool. Virology209:327–336. 2.Bacher, J. M., and A. D. Ellington.1998. Nucleic acid selection as a tool for
drug discovery. Drug Discovery Today3:265–272.
3.Back, N. K. T., M. Nijhuis, W. Keulen, C. A. B. Boucher, B. B. Oude Essink, A. B. P. van Kuilenburg, A. H. van Gennip, and B. Berkhout.1996. Reduced replication of 3TC-resistant HIV-1 variants in primary cells due to a proces-sivity defect in the reverse transcriptase enzyme. EMBO J.15:4040–4049. 4.Bahner, I., K. Kearns, Q. L. Hao, E. M. Smogorzewska, and D. B. Kohn.
1996. Transduction of human CD34⫹hematopoietic progenitor cells by a
retroviral vector expressing an RRE decoy inhibits human immunodeficiency virus type 1 replication in myelomonocytic cells produced in long-term cul-ture. J. Virol.70:4352–4360.
5.Bauer, G., P. Valdez, K. Kearns, I. Bahner, S. F. Wen, J. A. Zaia, and D. B. Kohn.1997. Inhibition of human immunodeficiency virus-1 (HIV-1) repli-cation after transduction of granulocyte colony-stimulating factor-mobilized CD34⫹cells from HIV-1-infected donors using retroviral vectors containing
anti-HIV-1 genes. Blood.89:2259–2267.
6.Beard, W. A., S. J. Stahl, H. R. Kim, K. Bebenek, A. Kumar, M. P. Strub, S. P. Becerra, T. A. Kunkel, and S. A. Wilson.1994. Structure/function studies of human immunodeficiency virus type 1 reverse transcriptase. Ala-nine scanning mutagenesis of an alpha-helix in the thumb subdomain. J. Biol. Chem.269:28091–28097.
7.Bebenek, K., W. A. Beard, J. R. Casas-Finet, H. R. Kim, T. A. Darden, S. H. Wilson, and T. A. Kunkel.1995. Reduced frameshift fidelity and processivity of HIV-1 reverse transcriptase mutants containing alanine substitutions in helix H of the thumb subdomain. J. Biol. Chem.270:19516–19523. 8.Bebenek, K., W. A. Beard, T. A. Darden, L. Li, R. Prasad, B. A. Luton, D. G.
Gorenstein, S. H. Wilson, and T. A. Kunkel.1997. A minor groove binding track in reverse transcriptase. Nat. Struct. Biol.4:194–197.
9.Berglund, J. A., B. Charpentier, and M. Rosbash. 1997. A high affinity binding site for the HIV-1 nucleocapsid protein. Nucleic Acids Res.25:1042– 1049.
10.Bordier, B., M. Perala-Heape, G. Degols, B. Lebleu, S. Litvak, L. Sarih-Cottin, and C. Helene.1995. Sequence-specific inhibition of human immu-nodeficiency virus (HIV) reverse transcription by antisense oligonucleotides: comparative study in cell-free assays and in HIV-infected cells. Proc. Natl. Acad. Sci. USA92:9383–9387.
11.Chen, H., and L. Gold. 1994. Selection of high-affinity RNA ligands to reverse transcriptase: inhibition of cDNA synthesis and RNase H activity. Biochemistry33:8746–8756.
12.Chen, H., D. G. McBroom, Y. Q. Zhu, L. Gold, and T. W. North.1996. Inhibitory RNA ligand to reverse transcriptase from feline immunodefi-ciency virus. Biochemistry35:6923–6930.
13.Ding, J., K. Das, and E. Arnold.1998. Structure and functional implication of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8Å resolution. J. Mol. Biol.284:1095–1111.
14.Dirani-Diab, R. E., L. Sarih-Cottin, B. Delord, B. Dumon, S. Moreau, J.-J. Toulme, H. Fleury, and S. Litwak.1997. Phosphorothioate oligonucleotides derived from human immunodeficiency virus type 1 (HIV-1) primer tRNALys3are strong inhibitors of HIV-1 reverse transcriptase and arrest
viral replication in infected cells. Antimicrob. Agents Chemother.41:2141– 2148.
15.Duzgunes, N., E. Pretzer, S. Simoes, V. Slepushkin, K. Konopka, D. Flasher, and M. C. de Lima.1999. Liposome-mediated delivery of antiviral agents to human immunodeficiency virus-infected cells. Mol. Membr. Biol.16:111– 118.
16.Evans, S. V.1993. SETOR: hardware-lighted three-dimensional solid model representations of macromolecules. J. Mol. Graphics11:134–138.
17.Giver, L., D. P. Bartel, M. L. Zapp, M. R. Green, and A. D. Ellington.1993. Selection and design of high-affinity RNA ligands for HIV-1. Rev. Gene
137:19–24.
18.Gunthard, H. F., J. K. Wong, C. C. Ignacio, J. C. Guatelli, N. L. Riggs, D. V. Havlir, and D. D. Richman.1998. Human immunodeficiency virus replica-tion and genotypic resistance in blood and lymph nodes after a year of potent antiretroviral therapy. J. Virol.72:2422–2428.
19.Huang, H., R. Chopra, and S. C. Harrison.1998. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science282:1669–1675.
20.Jaeger, J., T. Restle, and T. A. Steitz.1998. The structure of HIV-1 reverse transcriptase complexed with an RNA pseudoknot inhibitor. EMBO J.17:
4535–4542.
21.Jensen, K. B., L. Green, S. MacDougal-Waugh, and C. Tuerk.1994. Char-acterization of an in vitro-selected RNA ligand to the HIV-1 Rev protein. J. Mol. Biol.235:237–247.
22.Kensch, O., B. A. Connolly, H. J. Steinhoff, A. McGregor, R. S. Goody, and T. Restle.2000. HIV-1 reverse transcriptase-pseudoknot RNA aptamer in-teraction has a binding affinity in the low picomolar range coupled with high specificity. J. Biol. Chem.275:18271–18278.
23.Kew, Y., Q. Song, and V. Prasad.1994. Subunit selective mutagenesis of Glu89 residue in human immunodeficiency virus reverse transcriptase. J. Biol. Chem.269:15331–15336.
24.Kohn, D. B., G. Bauer, C. R. Rice, J. C. Rothschild, D. A. Carbonaro, P. Valdez, Q. Hao, C. Zhou, I. Bahner, K. Kearns, K. Brody, S. Fox, E. Haden, K. Wilson, C. Salata, C. Dolan, C. Wetter, E. Aguilar-Cordova, and J. Church.1999. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34⫹cells from the bone marrow of
human immunodeficiency virus-1-infected children. Blood94:368–371. 25.Konopka, K., N. Duzgunes, J. Rossi, and N. S. Lee.1998. Receptor
ligand-facilitated cationic liposome delivery of anti-HIV-1 Rev binding aptamer and ribozyme DNAs. J. Drug Target5:247–259.
26.Leung, D. W., E. Chen, and D. V. Goeddel.1989. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique1:11–15.
27.Lu, Y., V. Planelles, X. Li, C. Palaniappan, B. Day, P. Challita-Eid, R. Amado, D. Stephens, D. B. Kohn, A. Bakker, P. Fay, R. A. Bambara, and J. D. Rosenblatt.1997. Inhibition of HIV-1 replication using a mutated tRNALys-3 primer. J. Biol. Chem.272:14523–14531.
28.Osborne, S. E., I. Matsumura, and A. D. Ellington.1997. Aptamers as therapeutic and diagnostic reagents: problems and prospects. Curr. Opin. Chem. Biol.1:5–9.
29.Prasad, V. R., and S. P. Goff.1989. A novelin situcolony screening method to detect human immunodeficiency virus reverse transcriptase activity ex-pressed in bacteria. J. Biol. Chem.264:16689–16693.
30.Prasad, V. R., I. Lowy, T. De Los Santos, L. Chiang, and S. P. Goff.1991. Isolation and characterization of a dideoxyguanosine triphosphate-resistant HIV-1 reverse transcriptase expressed in bacteria. Proc. Natl. Acad. Sci. USA88:11363–11367.
31.Schneider, D. J., J. Feigon, Z. Hostomsky, and L. Gold.1995. High-affinity ssDNA inhibitors of the reverse transcriptase of type 1 human immunode-ficiency virus. Biochemistry34:9599–9610.
32.Sharma, P. L., and C. S. Crumpacker.1999. Decreased processivity of human immunodeficiency virus type 1 reverse transcriptase (RT) containing didanosine-selected mutation Leu747Val: a comparative analysis of RT vari-ants Leu747Val and lamivudine-selected Met184Val. J. Virol.73:8448–8456. 33.Strayer, D. S.1999. Gene therapy using SV40-derived vectors: what does the
future hold? J. Cell Physiol.181:375–384.
34.Tanese, N., V. R. Prasad, and S. P. Goff.1988. Structural requirements for bacterial expression of stable, enzymatically active fusion proteins containing the human immunodeficiency virus reverse transcriptase. DNA7:407–416. 35.Tuerk, C., and L. Gold.1992. RNA pseudoknots that inhibit HIV-1 reverse
transcriptase. Proc. Natl. Acad. Sci. USA89:6988–6992.
36.Tuerk, C., and L. Gold.1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science
249:505–510.
37.Tuerk, C., and S. MacDougal-Waugh.1993. In vitro evolution of functional nucleic acids: high affinity RNA ligands of HIV-1 proteins. Gene137:33–39.