Vol.62, No. 10 JOURNALOFVIROLOGY, Oct. 1988,p. 3718-3728
0022-538X/88/103718-11$02.00/0
Copyright © 1988,AmericanSociety forMicrobiology
Processing
of
gag
Precursor Polyprotein
of Human T-Cell
Leukemia
Virus Type
Iby Virus-Encoded
Protease
SEOK HYUN NAM, MINORU KIDOKORO, HISATOSHISHIDA, AND MASAKAZU HATANAKA*
Institutefor Virus Research, Kyoto University, Sakyo-ku, Kyoto 606, Japan Received 14 March1988/Accepted 13June1988
Thebiological activity encodedintheputativeproteasegene(pro)ofhuman T-cellleukemia virustypeIwas investigated by using a vaccinia virus expression vector. The 53-kilodalton gagprecursor polyprotein was processed into the mature pl9, p24, and p15gagproteins when thegagand protease-coding sequence was expressed under the control ofa vaccinia viruspromoter, suggesting that the protease may be synthesized through the mechanism of ribosomal frame shifting. The processing defect ofa protease mutant could be complemented by cointroductionofawild-typeconstructinto thecell,demonstratingthattheprogeneencodes thebiologically
active
proteasemoleculeswhicharecapable ofprocessingthegagprecursorpolyproteininvivo intrans.Astudyinvolvingtheuseofavarietyofmutantsconstructedinvitrorevealed that theproteaseconsists ofanonessential carboxy-terminal region andapart essential foritsactivity, includingtheputative catalytic residue,asparticacid.Furthermore,acluster of adenine residuespositionedattheoverlapping regionbetweenthegag andpro genes was shown to be involved in the ribosomal frameshifting event for the synthesis of
protease. To vmimicthe formation of the 76-kilodalton gag-proprecursor polyprotein formed by ribosomal slipping,thecoding framesof thegagandprogene wereadjusted. The processingof thegag-pro precursor polyproteindependedonanintactproteasegene,implyingthatacis-actingfunction of human T-cellleukemia virustype I proteasemaybenecessarytotriggertheinitialcleavageeventthatleadstotherelease ofprotease from theprecursorprotein.
Human T-cellleukemia virustype I(HTLV-I) is the first
human type C exogenous retrovirus associated with adult
T-cell leukemia(9, 31, 32, 50). HTLV-Ihas beenmolecularly cloned from the peripheral blood leukocytes of an adult
T-cell leukemia patient, andsubsequent characterization of
the viral genome has revealed that the HTLV-I genome is
composed ofgag,pol, env,andpX genes,theorderofwhich
is similartothat in known animalretroviruses. Theinternal
structuralproteins ofHTLV-I encodedin the gag geneare translatedasagag precursorpolyprotein of53 kilodaltons, which is subsequently processedinto p19,p24 and
p15,
the maturegagstructural proteins (8).Proteolytic cleavage of the gag precursorpolyprotein of murineand avianleukemogenicretroviruses hasbeenshown to be carried out by a highly substrate-specific virion-associatedprotease, whichplaysanimportantrolein infec-tivity of virus particles(17, 48). On the basis ofcomparison
ofthe amino acid sequence ofretrovirus-associated prote-aseswiththe DNA sequenceofclonedviralgenomes,it was concluded thatthe proteaseis encoded in the viral genome
itself(36,51-53).
We have molecularly cloned a novel full-length provirus
genomeofHTLV-Ifrom thevirus-producingcelllineMT-2 (24). This clone, designated as HTLV1C, harbors an open
reading frame (ORF) between the gag and the pol genes
which corresponds to the protease-coding region of other
retroviruses (26). This ORF had not been identified in the nucleotide sequence of the other isolates (33, 37). We have reported that amino acid sequences strictly conserved among other retroviral proteases are also found in the
possible protease-coding sequence of HTLV1C. Recently, another full-lengthDNA clone of
HTLV-I,
also containingthe ORF of theputative protease region, was independently
isolated from extrachromosomal closed-circular copies in
*Correspondingauthor.
chronically infected promyelocytic leukemia HL60 cells
(10). Both of the cloned viral DNAs harbor essentially the same ORFs forputative HTLV-I protease, except forafew base transitions. This strongly suggests that HTLV-I
en-codes it own protease. However, scarcity of the protease
associated with virions, together with unavailability of effi-cient virus-producing cells, has hampered our ability to examine thepropertiesofthe HTLV-Iprotease.
Vaccinia virus propagates in the cytoplasm exclusively,
and in infected cells a variety of genes are expressed in a regulated manner common toeucaryotic viruses and cells,
except for the absence of splicing involved in mRNA syn-thesis.Recently, the usefulness of vaccinia virusexpression
vectors has been established (39). Such vectors express foreign genes at high levels. We decided to adopt the vaccinia virus system to investigate the biological function andmode of expression of theHTLV-I protease gene.
In this studywe demonstrate that proteolytic cleavage of gag precursor polyprotein depends on HTLV-I protease, which is encoded in the putative protease gene region inferred from nucleotide sequencing. We also characterize the biosynthesis of the HTLV-I protease by using in vitro mutagenesis techniques.
MATERIALS AND METHODS
Materials. Allrestrictionendonucleases, as well as DNA-modifying enzymes, the M13 sequencing kit, and the phos-phorylated 8-nucleotide BgiII DNA linker were purchased from TakaraShuzoCo., Kyoto, Japan. Restriction endonu-clease BstXI and OxaNI werepurchasedfrom New England BioLabs, Inc., Beverly, Mass. Mung bean nuclease was obtained from Pharmacia, Uppsala, Sweden. Anti-p19 monoclonal antibody(GIN7), anti-p24 monoclonal antibody (NORI), anti-p15 monoclonal antibody (FR45),and partially purified HTLV-I virus proteins were kindly provided by Fujirevio Ltd., Tokyo,Japan. [a-32P]dCTPwas supplied by 3718
on November 10, 2019 by guest
http://jvi.asm.org/
PROCESSING OF HTLV-I gag POLYPROTEIN BY VIRAL PROTEASE Du Pont, NEN Research Products, Boston, Mass. Protein A
gold solution and its gold enhancement kit were purchased
from Bio-Rad Laboratories, Richmond, Calif.
General DNA method. Plasmid DNA was prepared by a
slight modification of the alkali-sodium dodecyl sulfate
(SDS) method of Birnboim and Doly (2). Restriction endo-nucleases and DNA-modifying enzymes were used as spec-ified by the manufacturers. All molecular-biological manip-ulations were carried out by standard methods (23). All DNA
transformationwere performed with competent Escherichia
coliJM109 cells. However, single-stranded DNAs for
site-specific mutagenesis were produced by using transformants
ofE. coli TG1 harboring subcloned M13 replicative-form
(RF) DNAs or infecting transformants of E. coli MV1184
harboringsubcloned pUC118 or pUC119 DNAs with helper
bacteriophageM13KO7 as described by Vieira and Messing (J. Vieiraand J. Messing, Methods Enzymol., in press). The
sequencing of single-stranded M13 virion DNAor
double-stranded plasmids was done by the chain termination method described by Sanger et al. (35).
Construction of plasmids. To construct p7.5gagN, we
isolated the 2.7-kilobase (kb) SstI-KpnI fragment of
HTLV1C that contains the 3' half of the long terminal repeat, gag, and pro and the 5' portion ofpol.This fragment wasdigestedwith
ApaI,
and a 2.3-kb fragment was purified andthenpartially digested withRsaIto remove the 3'halfof thelong terminalrepeat. FollowingtreatmentwithT4 DNApolymerase, a2-kbfragmentwasligatedwithSmaI-digested pProl8 (38), and then plasmid DNA from an ampicillin-resistant transformant was tested to selected the desired
plasmid by appropriate enzymedigestion.
To construct p7.5gagANS, p7.5gagN was digested to
completion with
StuI,
and the 1-kbStul
fragment, which contains the entirep15
codingsequenceandaportion of p24 and the protease-coding sequence, waspurified.
The 4-kbStuI
fragment harboring
thepUC
vectorsequence wasalso isolated for further experiments. The 1-kb fragment wasdigested completelywith Narl and treated withmungbean nucleasetodigestthe 5'protrudingendoftheNarlsite.
The
0.9-kbfragment wasligated withthe 4-kbStuI fragment of p7.5gagN, andplasmid p7.5gagANS was obtained.
The identical StuI fragments of p7.5gagM were used to construct
p7.5gagATS.
The1-kbStuIfragment
wasdigested
to completion with
TaqI
and then treated with the Klenow fragment ofE. coliDNA polymerase I to fill the end. Theenzyme-modified 0.6-kb fragmentwasrecovered and
ligated
with 4-kb StuI fragment as described
above.
The resulting plasmidwas designated asp7.5gagATS.
To construct the p7.5gagINBg, we
digested
p7.5gagN completely with OxaNI. After both ends have been filledwith Klenow fragments, linearized p7.5gagN was ligated with the 5'-phosphorylated synthetic octanucleotide
BglII
linker, 5'-CAGATCTG-3'.
Following complete
digestion
with
BglII,
the linearizedplasmid
was recircularizedby
ligationtoobtainthe
plasmid
p7.5gagINBg.
Insertion ofthelinkerDNAresultedintermination oftheprotease 10 amino
acids downstream ofthe OxaNI site.
To construct
p7.5gagAHO,
p7.5gagN waspartially
di-gestedwithHincll and
subsequently
digested
with OxaNItocompletion.
The 0.3- and 4.7-kbfragments,
harboring
aportion
oftheprotease-coding
sequence and the remainderof the
plasmid
including
thepUC
sequence,respectively,
were recovered from the gels. The 0.3-kb HincII-OxaNI
fragment wasfurther
digested
withHpaII
and then treated with Klenowfragmenttofillthe end.The,enzyme-modified
0.2-kb
fragment
wasligated
with the 4.7-kb HincII-OxaNIfragment of p7.5gagN, which was previously treated with
Klenow
fragment,
to producep7.5gagAHO.
Strategies
forconstruction
of in vitro-mutagenizedplas-mids were as follows. Plasmids p7.5gagfsl9AspI and
p7.5gagfsl9AspII
were constructed by using p7.5gagfsl9, p7.5gagNAspI, and p.7.5gagNAspII. The 0.6-kb HincII-EcoRIfragments
harboringthe in vitro-mutagenizedprote-ase-coding
sequence were purified from p7.5gagNAspl orp7.5gagNAspII.
Thesefragments
were used to replacethe
corresponding
region
of p7.5gagfsl9 to obtainp7.5gagfsl9Aspl
andp7.5gagfsl9AspII.
To constructp7.5gagfsl9AOE, p7.5gagfsl9AspI
wascompletely
digested
with OxaNI and EcoRI and then treated with the Klenow fragment ofE. coliDNApolymeraseI tofill bothends. The 4.7-kb OxaNI-EcoRI
fragment
was circularized by self-ligationtoproducep7.5gagfsl9AOE. This truncatedgag-profusion
protein
may have 26carboxy-terminal
amino acids contributedby pProl8.
Anovelrestrictionenzymecleavage site,KpnI, createdat
amino acid 125 ofthe protease was used to truncatethe 3'
region
oftheprotease-coding
sequenceinp7.5gagfsl9AspII.After complete digestion with KpnI, the 4.6-kb KpnI frag-ment of
p7.5gagfsl9AspII
was recircularized to generatep7.5gagfsl9AKp.
Thistruncatedgag-profusionprotein
mayhave 32
carboxy-terminal
amino acids contributedby
pProl8.
In vitro mutagenesis.
Oligonucleotide-directed
in vitromutagenesis
wasperformed by using
amutagenesis
kitsupplied by
AmershamInternational,
Amersham,England,
as described
by Taylor
et al. (43), except that the reaction volume was reduced to half of theoriginal
volume. Todisrupt
the essential amino acid sequence forproteolytic
cleavage
locatedbetweenp19andp24
junction,
weexcised a 0.6-kb BamHI-PstIfragment, harboring
the entirep19
coding
sequence and the 5'portion
of thep24 coding
sequence,from
p7.5gagN
and thenligated
it withM13mpll
RF DNA which had been
previously digested
withBamHI andPstI.Site-specific
mutagenesis
wasperformed by
using
the standard
protocol specified
by
themanufacturer,
withthe 37-mer
oligonucleotide
5'-TGAGCCTACGGCGCCCC CAAGTCCTTCCGTCATGCAT-3'. One base deletion at nucleotide1179,
together
withonebaseinsertionat nucleo-tide1194,
gave risetothelocalized frameshifting
atthep19
-
p24
junction region,
with simultaneous amino acidse-quence
changes
from Pro-Gln-Val-Leu-Pro to Pro-Pro-Ser-Pro-Ser. SubclonedM13mpll
RF DNA that contained themutagenized
sequencewasdigested
withBamHI andNcoI,
and then the 0.5-kb
fragment
waspurified.
Thisfragment
was
ligated
withthe4.1-kbfragment
ofp7.5gagATS,
whichhad been
digested
completely
with BamHI andNcoI,
toproduce
p7.5gagATS37.
To construct
p7.5gagfsl9,
the 0.9-kb SmaI-EcoRIfrag-ment
harboring
theentireprotease-coding
sequence and the 3' half of thep15 coding
sequence was excised fromp7.5gagN
and then insertedatthecorresponding
restrictionenzymesites of
M13mplO
RFDNA.Mutagenesis
was car-ried out with the 19-meroligonucleotide
5'-ACACCCAAA
GAAACTCCAT-3'. Direct
sequencing
ofthemutagenized
region
showed the creation ofa new DNAsequence, AAAGAAAC,
insteadofAAAAAAC
atsix consecutive adenineresidues
positioned
atthe3' terminalregion
of the gag gene. The 0.2-kb BstXI-HincIIfragment
was excised from sub-clonedM13mplO
RF DNAharboring
themutagenized
target
DNA
by
complete
restriction enzymedigestions
and thenligated
with the 4.8-kbfragment
ofp7.5gagN,
which hadVOL.62, 1988 3719
on November 10, 2019 by guest
http://jvi.asm.org/
3720 NAM ET AL.
been partially digested with Hincll and subsequently di-gested with BstXItocompletion.
The 0.9-kb SmaI-EcoRI fragment described above was also inserted into the corresponding enzyme site of M13mpll RF DNA, and in vitro mutagenesis was carried out with using5'-CCTATGGAGAATATTGGGTGTG-3' as amutagenic oligonucleotide. Asaresult ofmutagenesis, the newDNAsequencerepresented byAATATTCwascreated instead of AAAAAAC, with simultaneous creation of a novelrestrictionenzymecleavagesiteofSspI ofsix consec-utive adenine residuespositionedatthe 3' terminalregionof thegaggene. To construct p7.5gagfs22, we introduced the mutagenized DNA fragment into p7.5gagN by using a
pro-cedure identicaltothat used for construction ofp7.5gagfsl9. To change the amino acidat position 64 ofthe protease fromasparticacidtoglycine,weexcisedthe0.6-kb HincII-EcoRI fragment harboring the protease-coding sequence
from
p7.5gagN
and ligated it with pUC118 linearized by previous HincII and EcoRI digestion. Mutagenesis was performed by using 5'-AAGCTCTACTAGGTACCGGAG CAGACATGACAGT-3' as a mutagenic oligonucleotide. The mutagenized clone was judged by the creation of a restriction enzyme siteKpnIatthe novel glycinecodon. A similarprocedurewasadoptedtoexchangethe amino acidatposition125 fromasparticacidtoglycine.The 0.6-kb HincII-EcoRIfragmentdescribed above was insertedat the corre-sponding restriction enzyme sites ofpUC119. Mutagenesis was performed by using 5'-GTTTTTGGTACCAACTAGG-3' as a mutagenic oligonucleotide. The mutagenized clone wasjudged bythesimultaneous creationof the novel restric-tion enzyme cleavage site of KpnI at the novel glycine codon. To construct p7.5gagNAspl or p7.5gagNAspII, which contain the newly created glycine residues at amino acidpositions 64 and 125 ofthe protease, respectively, we purifiedthe0.3-kb HincII-OxaNIfragmentsfromsubcloned pUC118 or pUC119 DNA by complete restriction enzyme digestions and then ligated them to the 4.7-kb fragment of p7.5gagN,whichhadbeenpartially digestedwithHinclI and subsequently digestedto completionwith OxaNI.
Recombinantvaccinia viruspreparation.Vaccinia-HTLV-I recombinant plasmids for recombinant vaccinia virus were constructedasfollows. Plasmid p7.5gagNwasdigestedwith HindIII and EcoRI, and the 2.3-kb fragment harboring the entiregag andprotease-codingsequence includingthe
pro-moterofthe 7.5Kgene was purified. The large fragment of E. coli DNA polymerase I (Klenow) was used to fill both ends of this fragment. To flank the fragment with the hemagglutiningene of vaccinia virus, we inserted the
frag-mentinto pHA13 which hadbeen linearized with NruIand subsequent treatment of Klenow fragment to fill the ends (38). Candidatecloneswere tested fortheorientation of the insert,andthedesiredplasmidwasdesignatedaspHAgagN. The 2-kbHindIII-EcoRIfragment harboring the entiregag gene, including the truncated protease-codingsequenceand the 7.5K gene promoter, was excised from p7.5gagATS. After treatment withthe Klenow fragment ofE. coli DNA polymerase I,thisfragmentwasinserted in totheNruI site ofhemagglutiningene of vacciniavirusasdescribed above. The resultant plasmid was designated aspHAgagATS. The detailedprocedure for generation and purification of recom-binantvaccinia virus was described by Shidaetal. (39).
DNA transfection. CV-I cells for transfection of plasmid DNA were maintained in Eagle minimal essential medium supplementedwith10% fetalcalfserumat37°C in humidified
airwith5%CO2. Transfection of plasmid DNAs for transient expressionwas performedasdescribed (6). Briefly,2 x 105
to 3 x 105 cells per 35-mm dish were preinfected with wild-type vaccinia virus at 30 PFU per cell and adsorbed for 1 h at 37°C. Then, 10
jig
of calcium phosphate-precipitated recombinant plasmid DNA was added. After a30-min incu-bation at room temperature, fresh medium was added. After a 4-h incubation at37°C, the medium was replaced by fresh medium; incubation was continuedovernight.Viral protein analysis. At 20 h posttransfection with recombinant plasmid DNA, CV-I cells were washed twice with phosphate-buffered saline and then solubilized in SDS-sample buffer by heating for 3 min at 95 to 100°C. Solubilized proteins were separated by SDS-polyacrylamide gel electro-phoresis (12% polyacrylamide) and transferred onto a nitro-cellulose filter as described (45). The blots were blocked in fetal calf serum at 4°C overnight and then probed with the
anti-gag monoclonal antibodies appropriately diluted with 3% bovine serum albumin solution containing 20 mM Tris (pH 8.0) and 150 mM NaCl. After the primary antibody reaction had continued at room temperature for 3 h, the secondary antibody reaction, with rabbit anti-mouse
immu-noglobulin G antibody, was performed under the same conditions. Finally, the protein A-gold reaction and subse-quent enhancement were performed as specified by the
manufacturer; the gag-related proteins expressed in CV-I cells transfected with the recombinant plasmids were re-vealed.
RESULTS
Processing of gag precursor polyprotein in a recombinant vaccinia virus expression vector system. The2-kb RsaI-ApaI fragment harboringthe entiregag and pro ORF was purified
andintroduced downstreamofthe promoterofthe vaccinia
virus7.5K gene, which is expressed atboth earlyand late times in the vaccinia virus infectioncycle (3). The
construc-tion ofplasmid p7.5gagN is outlined in Fig. 1 and is de-scribedin Materials and Methods.
The plasmid was introduced into vaccinia virus-infected
CV-I cells by using the calcium phosphate coprecipitation technique, and thentransientexpression ofgag gene
prod-uctsinCV-I cellswasexaminedbyWesternimmunoblotting analysis with anti-gag monoclonal antibodies.
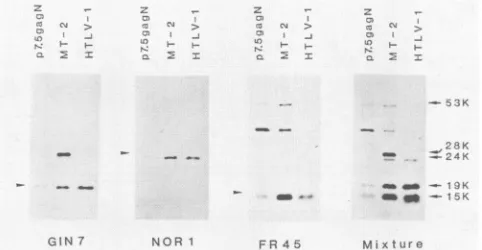
The 19-, 24-, and 15-kilodalton proteins (19K, 24K, and 15K proteins, respectively) wererecognized by ananti-p19 monoclonal antibody (GIN7), an anti-p24monoclonal anti-body (NORI),and ananti-p1S monoclonal antibody (FR45),
respectively (Fig. 2). Small amounts ofa 53K proteinwere
recognizedby FR45, which is the uncleaved gag precursor
polyprotein. A 36K protein, which is considered to be an
intermediate precursor protein, was also detected. All the gag proteins transiently expressed from plasmid p7.5gagN
comigratedin SDS-polyacrylamide gel electrophoresis with
the corresponding HTLV-I gag proteins in extracts of
HTLV-I-producing MT-2 cells and in isolated viruses. The 28K protein observed in MT-2 cells is not the processed
product ofgag precursor polyproteins, but is the gag-pX
fusionprotein consisting ofp19, part of p24, and a peptide from the pX region, and is encoded by the defective provi-ruses (12). These results show that the cloned gag and pro
ORF
region
is enough to synthesize all gag proteinscor-rectly processed fromtheprecursor polyprotein. As a matter ofconvenience, subsequent Western blot experiments were carried outby usingthemixed anti-gag monoclonal
antibod-ies GIN7, NORI, and FR45.
Proteolytic activity residues within the putative gene prod-uctof theproORF. To demonstrate that the processing of gag
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
PROCESSING OF HTLV-1 gag POLYPROTEIN BY VIRAL PROTEASE
HT LVI C
53 K
pro ~~~env
gag ro
Pl9
I
P24 p5Ptt
t
t
t
: Ut 1) cn D
In
t
13*
l-A31-4,_
--~~V o23 K _ -- 1 K
o l -1 K
GIN7
RsaI- Apa I 2 Kb fragment
T4 polymerase
Sm-I
;gation
H;ndmIl
EcoR I
FIG. 1. Schematic representation of the HTLV-1 genome and
construction of p7.5gagN. Thestrategyusedtoinsert the entiregag
andprotease-codingsequence into the pPro18expressionvectoris outlined.Symbols: c.gag,pol,env,and pXsgenesof HTLV-I; _, 7.5Kgenepromoterof vacciniavirus; ,gagand
protease-codingsequence;circle, pUC18 DNAsequence; *,positions of the HTLV-I protease-mediated processing sites;-*, direction of
tran-scription.
precursorpolyprotein observedinthevaccinia virus
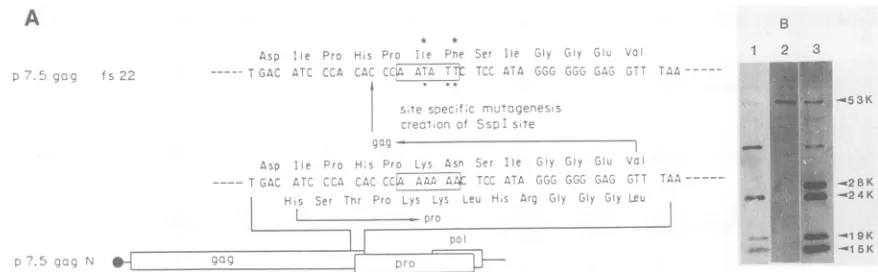
expres-sionsystemisaccomplished byHTLV-Iproteaseencoded in nucleotide sequences within thepro ORF, we deleted sev-eral regions ofprotease-coding sequence(Fig. 3A).
Adeletion from the Narn sitetotheStilT site(p7.5gagANS) did not abolish the correct processing of gag precursor polyprotein (Fig. 3B, lane 2). However, processing was blocked when the deletion was extended to the TaqI site (p7.5gagATS), located atthe 5' region of thepro ORF (lane 3), and the gag precursor polyprotein (Pr53gag) accumu-lated. The minor immunoreactive bands of lower molecular mass were consideredto be degradedgagproducts. Trans-lational termination of the protease as aresult ofthe inser-tion of an 8-nucleotide BglII linker at the OxaNI site (p7.5gagINBg) had no effect on correct processing of the
53Kgagprecursorpolyprotein (lane 4). Theseresultsimply
thata region spanning nucleotide positions 2228 to 2495 of theproORFisimportanttotheprocessingofPr53gag into the mature gag products. By contrast, the region down-streamfrom the OxaNI site ofpro ORF is notessential for
proteolytic function. This is consistentwith the observation that the region from nucleotides 2228 to 2495 encodes the
NOR 1 FR45 M x tu re
FIG. 2. Expressionand processing of gag precursorpolyprotein in CV-1 cells by using vaccinia virus-derived expression vector. Plasmid p7.5gagN was constructed as shown in Fig. 1. Proteins expressed fromthe plasmid were analyzed on an SDS-12% poly-acrylamide gel andelectrophoretically transferred onto nitrocellu-lose filters. Transient expression of gag-related protein were ana-lyzed by Western blotting analysis with anti-p19 monoclonal antibody (GIN7),anti-p24 monoclonal antibody (NORI), and anti-p15 monoclonal antibody (FR45), or their mixtures. Arrows indi-cated the mature gag proteins expressed from p7.5gagN. Virion-associated gag proteins (HTLV-I) and those synthesized in the chronically infectedcells (MT-2) werealsoanalyzed toconfirmthe authenticity of the mature gag proteins expressed from the p7.5gagN.
amino acid stretch that is conserved among the HTLV
family of retroviruses.
To further examine the involvement of conserved amino acid stretches in the cleavage of gag precursor, we con-structed plasmid p7.5gagAHO, which contains the entire
protease-coding sequence, except that a region harboring one of the conserved amino acid stretches was deleted (Fig. 3A). The major immunoreactive band encoded by
p7.5gagAHO was the 53K protein, although nonspecifically degraded proteins were also observed (Fig. 3B, lane 5). These resultsfurther support theassignmentof the pro ORF regionasthe coding sequence for HTLV-I protease.
The processing defect of the pro ORF deletion mutant is complemented byactiveprotease. Toobtain evidence that the
intact proteasemolecule directly cleaves the gag precursor
polyprotein, we needed to determine whether the gag pre-cursorpolyproteinencodedbythe proORF deletion mutant was able to be processed in trans by wild-type protease in vivo. Thevaccinia virusexpression vector system is suitable for this kind ofstudy,becausetwoindependentrecombinant DNAscanbecointroduced byusingthe recombinant vacci-nia virus as a helper virus. The detailed strategy for con-struction ofplasmidsis shownschematicallyinFig.4.Asthe efficient donor of the active form of protease,arecombinant vaccinia virus expressing the protease was constructed by usinghemagglutinin gene-mediated homologous
recombina-tion as described in Materials and Methods. The resultant
recombinant vacciniavirus, WRgagN,
harboring
the gag and pro ORF, produced processed gag products identical to those synthesized in MT-2 cells (Fig. SA, lane 4). For the control experiments, recombinant vaccinia virus harboringthe gag ORF alone was constructed and
designated
WRgagATS.No correctprocessingoccurred in cells infected withWRgagATS (lane 5). The
p7.5gag/TS
plasmid,
encod-ing onlythe 53K gag precursor,wasmutagenized
in vitrotodisrupt the essential amino acid sequence for
proteolytic
cleavage located between
p19
andp24.
The resultantplas-mid,
p7.5gagATS37,
encoded a novelp19-p24
junction
se-VOL.62, 1988 3721
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.71.277.71.407.2] [image:4.612.317.558.75.200.2]3722 NAM ET AL.
Hindm_ EcoRI H:ndm coR I
p7.5gagN 5a
EcoRI Nru I EcoRI
Hindlm Sal Hindm Hind m
2.3Kb HA 2Kb
fragment HA 13 fragment
klenow | kNenow/ |
|NruI l
ligoation
proc e s s ng
O O x 0 X
1 2 3 4 5 6
_- _ _- g3K
WRggATS
I
0 28K
-24K
-.-_19K
-1 5K
FIG. 3. Plasmidsencodingthe truncated proteasemolecules of HTLV-I anditsexpression. (A)Mapofpartofthe HTLV-Igenome
isgivenatthetopof thefigure. The nucleotide number indicatedin
thisfigurecorrespondstothatreported bySeikietal.(37). Several deletionmutantsofprotease-codingsequence wereconstructedas
described inMaterials and Methods. Symbols: 0, 7.5Kgene
pro-moter of vaccinia virus; izz, gag coding sequence ofHTLV-I;
,protease-codingsequence; = ,reversetranscriptase coding
sequence ofHTLV-I; V, insertion of8-nucleotidephosphorylated
BgIII synthetic linkerat OxaNI site; S,twoconservedamino acid stretches betweenthe retroviralproteasesand thecellularaspartyl
proteases; C, short amino acid sequences conserved only among
HTLV-family retroviral proteases. Abbreviations: Rs, RsaI; Sm, SmaI; Nc, NcoI; Ps, PstI; St, StuI; Bx, BstXI; Hc, HincII; Tq, TaqI; Hp, HpaII; Ox, OxaNI; Na, Narl; Ap, ApaI. (B) Western blotting analysisofgagproteins expressedfrom deletionmutantsof protease-coding sequence. Evidenceforprocessing is indicatedby
signals, represented by 0 (processing positive) or x (processing
negative). The mixed anti-gag monoclonal antibodies of GIN7, NORI, and FR45 were used to detect the HTLV-I gag-related proteinsfrom celllysates.
quence Pro-Ser-Pro-Ser, instead ofthe wild-type
Gln-Val-Leu-Pro(Fig. 4B). Thisallowed the processedgagproducts
directedbyp7.5gagATS37 and those derived fromWRgagN to be distinguished.
Western blotting analysis showed that p7.5gagATS37 di-rectedthesynthesis ofa53Kgagprecursorpolyprotein with
no evidence of the natural gag proteins (Fig. SA, lane 3).
Whenp7.5gagATS37wasintroduced into CV-I cells infected with WRgagN, a new 43K protein was identified, although
p19, p24, andp15derivedfrom WRgagNwereevident (lane
1). This protein was no longer detected when WRgagATS
B
p7.5gagATS37- ACG GCG CCC CCA AGT CCT TCC
GTC----Thr Ala Pro Pro Ser Pro Ser Val
* *
sitespeC;fiCmutogenesis
_ _ACG GCC CCC CAA GTC CTT CCA GTC----Thr Ala Pro _in Val Leu Pro V
p7.5gagATS < pi9 P24 P15
cx LX C
FIG. 4. Schematic representation ofplasmid constructions for establishment of enzyme-substrate reaction system in vivo. (A) Constructionofplasmidvectorsfor transfer ofthegagand protease-coding sequence of HTLV-I into vaccinia virus. Symbols: Ez 1, hemagglutinin gene of vaccinia virus; , gag and
protease-codingsequenceofHTLV-I; _,7.5Kgenepromoterderivedfrom vacciniavirus; the direction oftranscription. (B) Schemefor
con-struction of plasmid p7.5gagATS37. Symbols: 0, 7.5Kgene
pro-moterof vacciniavirus; ,truncatedprotease-codingsequence;
,5'portionofreversetranscriptasegene;0or0,insertionor
deletion of nucleotide causedbyoligonucleotide-directed site
spe-cific mutagenesis; *, exchanged amino acid residues caused by site-specificmutagenesis.
was used asthe helpervirus(lane 2). We presume that the 43K protein represents a processed mutant gag precursor polyprotein that contains p19 plus p24 but not p15. The identity of the 43K protein was further examined by using individualanti-gag monoclonal antibodies. As expected, the 43K protein was recognized by p19 monoclonal anti-body (GIN7) and anti-p24 monoclonal antibody (NORI), (Fig. SB, lanes 1 and 2). Anti-p1S monoclonal antibody (FR45) failed to recognize this protein (lane 3). Thus, we conclude that HTLV-I protease encoded inthepro ORF is
ableto process thegagprecursorpolyprotein intrans.
Importance ofasparticacid residuesin HTLV-I protease.
A A
B
J. VIROL.
I
I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.64.301.72.417.2] [image:5.612.325.556.80.479.2]PROCESSING OF HTLV-I gag POLYPROTEIN BY VIRAL PROTEASE
7T. '
B
6 1 2 3 4
- - -53K
-28K _
-24K _
- -19K
-ww 15K
A
s{.tt-4t';4
~~~~~~~~~~~~~~~~~~~~~7.5
r i A^9 [image:6.612.65.299.74.291.2] [image:6.612.322.557.75.410.2]A~~~~~~~
pp.
errr
{| e es s
_ r-T-^--A---T----
---*~~~~=<7.~ ~~~~~~~~~~~~~~~~~~~~~~~~5 g gr4
Leu a2I Lsp Tr L ys
|
s
e ;-i:+-7enes;s |ce3 , otf K-rl 5seITj T C-fT A A
L.Pi .,a CIy Tb,r L.ys ESsr.;T.
WPI.. ..::..
B
p 5 "rN hSF I
1
2
3
4
FIG. 5. Identificationand characterization of43Kprotein
gener-ated by the enzyme-substratereaction in vivo.(A)Identificationof
the 43K processedprotein from themutantgagprecursor
polypro-tein byanintransaction of HTLV-Iprotease.The substratedonor,
p7.5gagATS37, was expressed in CV-I cells by using wild-type
vaccinia virusasthe helpervirus (lane3)or wasexpressed byusing
recombinant vacciniavirus harboring the protease-codingsequence
(WRgagN)asthe helper virus (lane1). Aprotease-coding
sequence-defective recombinantvaccinia virus (WRgagATS)wasalso usedas
thehelper virusfor the control experiment (lane2). Thecellextracts
wereseparatedon anSDS-12%polyacrylamide gel, andsubsequent
Westernblotting analysis wascarriedoutby using mixedanti-gag
monoclonalantibodies of GIN7,NORI, and FR45. Production of the active form ofthe HTLV-I protease molecule from recombinant vaccinia viruswasjudged by thecorrectprocessingeventsof53K
gag precursorpolyproteins (lanes 4and 5). MT-2cell lysate was
usedtoshowthecorrectmobilities of authenticmaturegagproteins
ofHTLV-I inSDS-polyacrylamide gel electrophoresis (lane 6). (B) Characterization of the antigenicity ofthe 43K protein. Proteins expressed from p7.5gagATS37 with WRgagN as the helper virus were separated by SDS-polyacrylamide gel electrophoresis (12%
polyacrylamide). SubsequentWesternblotting analysiswascarried
outbyusingtheindividual anti-gag monoclonalantibodies, GIN7, NORI, andFR45, respectively(lanes 1, 2, and 3),or amixture (lane
4).Arrowsindicate 43Kproteins reacted withGIN7orNORI. The possible processingeventof themutantgagprecursorpolyproteinis schematically shown at the top of the figure. x indicates the
disruptionoftheconservedaminoacidsequenceforspecific prote-olysis. ,Vaccinia virusgenome.
Alignment of the amino acidsequencesofHTLV-Iand other retroviral proteasesrevealedthataminoacid sequences are strictlyconserved intwostretches,L-L-D-T-G and I-I-G-R-D(16, 26).We haveshownwiththeevidenceabove that the region containing these conserved amino acid sequences plays an important role in the expression of proteolytic activity (Fig. 3).
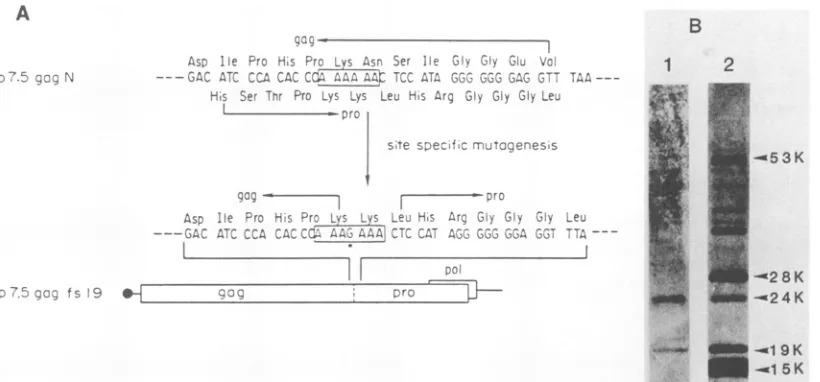
Thefact that L-L-D-T-G is found in thegroupofaspartyl
proteases, which includes pepsin and others, suggests that the asparticacid residue located in this sequence might be the catalytic center for HTLV-I protease. To identify the role of the aspartic acid positioned at residue 64 in the
catalytic activity of HTLV-I protease, we performed
site-specific mutagenesistochangethe aminoacid fromaspartic acidtoglycine.Theresultantmutantplasmid,p7.5gagNAspI
5 3 K
28K 2 24K
_19K
__~ -1 5K
FIG. 6. Inhibition of proteolytic activity of HTLV-I protease resulting from the exchange ofthe putative catalytic residues by in vitro mutagenesis. (A) Oligonucleotide-directed site-specific muta-genesisforgenerating p7.5gagNAspl andp7.5gagNAspII. Symbols:
0,7.5K gene promoterof vacciniavirus;0, twoconserved amino
acidstretchesbetweentheretroviralproteasesand cellularenzyme
groupofaspartylproteases;0, short aminoacid stretch conserved onlyamongHTLV-familyretroviral proteases;*, nucleotide
tran-sitionscausedbyoligonucleotide-directed site-specific mutagenesis; *, exchanged aminoacid residuesresulting from thetransitions of nucleotides; -, pUC DNAsequence.(B) Expression of wild-type
andmutantplasmids. Theextracts, preparedfromcellstransfected
with p7.5gagN (lane 1), p7.5gagNAspl (lane 2), and p7.5gagNAspII (lane 3), were analyzed on an SDS-12% polyacrylamide gel, and then Western blotting analysis was carried out by using mixed anti-gagmonoclonal antibodies ofGIN7, NORI, andFR45. MT-2 cell lysate was used as the marker of the authentic mature gag
products (lane4).
(Fig. 6A), directed the synthesis of the 53K gag precursor polyproteinbutnootherspecifically processed proteins (Fig. 6B, lane 2), providingfurther evidence that HTLV-I prote-ase belongsto thegroupofaspartyl proteases.
Inthe retroviral proteases of the HTLV family, an addi-tional short sequence is conserved between the two well-defined conserved amino acid sequences (89). This short
sequence, L-V-D-T (HTLV-I protease sequence positions
123 through 126), partially resembles that surrounding the
catalytic residue, asparticacid64. To examine thebiological significance ofthe additional short conserved sequence in the catalytic activity of the protease, we performed site-specific mutagenesis out to change the aspartic acid to
glycine at amino acid 125 (Fig. 6A). The mutant plasmid
1 2 3 4 5
_ _ -_.o _
A
43K-VOL.62, 1988 3723
on November 10, 2019 by guest
http://jvi.asm.org/
3724 NAM ET AL.
A
B
&.
..._...
_ -19K
FIG. 7. Abolition ofthe correctprocessingofgag precursorpolyprotein byblockingthe frameshifteventfor thesynthesisofHTLV-I
protease. (A) Oligonucleotide-directed site-specific mutagenesis for generating p7.5gagfsl9. The box encloses six consecutive adenine residues presumably involved in a frameshift event forsynthesis ofHTLV-l protease. Symbols: *, nucleotide transitions resultingfrom oligonucleotide-directedsitespecificmutagenesis;*,exchangedaminoacid codoncausedbytransition ofnucleotides;0,7.5Kgenepromoter ofvaccinia virus.(B)Theextracts,prepared fromcellstransfectedwithp7.5gagN(lane 2)andp7.5gagfs22 (lane 1),wereanalyzedon an SDS-12% polyacrylamidegel, andsubsequent Westernblotting analysiswas carriedoutwithmixedanti-gagmonoclonalantibodies.gag-related proteins synthesized in MT-2 cells werealsoanalyzedunder the sameconditions(lane 3).
p7.5gagNAspII also directed the synthesis ofonly unproc-essed 53K gag precursor polyprotein (Fig. 6B, lane 3),
suggesting anessential rolefor the asparticacid at position 125 in the HTLV-I protease function. We ascertained by
nucleotide sequencing that no unwanted nucleotide transi-tions occurred in the mutant plasmids, p7.5gagAspI and p7.5gagNAspII, during the in vitro mutagenesis procedure
(data not shown).
Possible model for biosynthesis of HTLV-I protease. We have suggested two possible mechanisms for synthesis of HTLV-I protease that explain the absence ofan initiation
methioninecodonatthe 5' terminus of the pro ORF and the
differenceinitsreadingframefromthatof the gag gene(26): one is that the mature mRNA encoding the protease is generatedby splicing, and the other is that the gag termina-tion codon is not read owing to an upstream frameshift. Correct processing ofthe gag precursor polyprotein in the
vaccinia virus expression vector system strongly suggested the frameshift mechanism, because vaccinia virus propa-gatesexclusivelyin thecytoplasm of the host cells and does not possess its own machinery for splicing (46). Recently,
ribosomal frame shifting in mouse mammary tumor virus has been demonstrated (11, 15). Toobtainmore direct evidence for frame shifting, we introduced mutations of three of the sixconsecutive adenines(nucleotide positions 2064through 2069) located in the overlapping region of the gag and pro
ORF. The mutant plasmid obtained was designated p7.5gagfs22 (Fig. 7). These consecutive adenine residues were expected to be involved in putative frame shifting by analogy with other retroviruses such as bovine leukemia virus. In mutant p7.5gagfs22, three A-to-T transitions were generated without disruption of the phase of the gag gene (Fig. 7A). The result of Western blotting analysis is shown in Fig. 7B. Processing of gag precursor polyprotein was no longer observed (lane 2), indicating that these residues are essential for generation of active protease. One may specu-latethat the mature protease is generated as follows: a frame shift gives rise to a gag-pro fusion protein with protease
activity,which is expected to be 76 kilodaltons in size. This gag-pro fusion protein is rapidly cleaved by its own or a cellular protease to generate more free protease molecules that can act in trans on the 53K gag precursor polyprotein. Tosubstantiate this scheme, it is important to demonstrate the presence of the 76K precursor polyprotein and then to
identify which protease is involved in its cleavage. In HTLV-I-producing cells, the 76K protein is hardlydetected, probably because of inefficient frame shifting and/or rapid processing. To mimic the naturally occurring ribosomal frameshifting event, we introduced a 1-base insertion to align the coding frames of the gag and pro genes. The insertion was at the consecutive adenine residues located in the 3' terminal region of gag gene of the protease-negative or protease-positive plasmids (p7.5gagNAspl, p7.5gagNAspII, andp7.5gagN) (Fig. 8 and 9). The resultant plasmids were namedp7.5gagfsl9Aspl p7.5gagfsl9AspII, andp7.5gagfsl9.
Although 76K proteins immunoreactive with anti-gag mono-clonal antibodies were produced in the cells transfected by p7.5fs19AspI andp7.5gagfsl9AspII (Fig. 9B, lanes 1 and 2),
p7.5gagfsl9directed andsynthesisof theprocessedproducts ofp19 and p24 (Fig. 8B, lane 1). Thep15(or related peptide) wasnot detected by theanti-p1S monoclonal antibody used in thisstudy,probablybecause of theconformationalchange induced bythe mutation.
These results canbe interpreted to mean that the HTLV-I-encodedprotease cantrigger the firstcleavageeventforits own release form the 76K gag-pro precursor form. How-ever, we cannot exclude the possibility that acell protease
initially activates the viral protease by cleaving it from an inactive precursor. Toverifythat the 76Kproteinband isnot artifactually cross-reacting with anti-gag monoclonal
anti-bodies,weconstructedplasmids with serial truncationsfrom
the 3' terminus of the pro gene (Fig. 9A). The size of the programmed products, 68K and 65K (directed by
p7.5gagfs19AspIAOE and p7.5gagfsl9AspII/Kp, respec-tively; Fig. 9A and B, lanes 3 and 4), were as expected for the truncatedgagpolypeptides. These results confirmed that 76Kprotein observed is the gag-pro precursorpolyprotein.
DISCUSSION
Retroviral proteases responsible for processing gag pre-cursorpolyproteins playanimportant role in the production ofinfectious virus particles. In a previous communication, we reported that the HTLV-I genome cloned from a virus-producingcell line MT-2 harbored an ORFfor the putative HTLV-I protease (26). In this study we examined the
biological function and properties ofHTLV-I pro ORF by
using the vaccinia virus expression vector. This system is
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.80.519.73.209.2]PROCESSING OF HTLV-I gag POLYPROTEIN BY VIRAL PROTEASE
A
B
As5 Iee P .Hrs _rc vs Sr Ser ive J
HiS e5 Tr. Pro -VS LYS Leoh-Ars
irg
'Vi' Le,sTe- sse:'erncc.e'
_ _
F
cAs rton ~s Dlr-- L"vs _ , r G ; _e
C
1 2
-53K
I
-24
*-p
"i9K 15 KFIG. 8. (A) Correctly processed gag proteins were generated from the gag-protease fusion protein. The box encloses six consecutive adenine residues of possible frameshift site for synthesis of gag-pro fusion protein. Symbols: 0, one nucleotide insertion introduced by oligonucleotide-directed site-specific mutagenesis; 0,junction between the gag and protease-coding sequence in the fusion protein. (B) Proteins expressed fromp7.5gagfsl9was separated on an SDS-12%polyacrylamide gel, and thenWesternblottinganalysis was carried out with mixedanti-gag monoclonal antibodies (lane 1). MT-2 celllysate was used as the marker to show the migrationpatternof the authentic mature gagproteins ofHTLV-I(lane 2).
characterized by a high frequency of DNA transfection and
by the capacity toefficiently express foreign gene products
in eucaryoticcells.
Weusedacombination oftwo approaches todemonstrate thattheproORFis the biologically functional protease gene
of HTLV-I. The first method involved deletion of appropri-ate regions in the putative protease-coding sequence. The results revealed that deletion of a region containing the
amino acidsequences strictlyconserved among all retroviral proteasesresulted inlossof proteolyticactivity (Fig. 3). The secondmethod is theestablishmentof an enzyme-substrate reaction system in vivo. We showed that the processing
defect of thegag precursorpolyprotein, directed bythe pro
ORF deletion mutant, could be complemented by cointro-duction ofa recombinant DNA constructed to have wild-type proORF. Thisresultprovides direct evidence that the proORF isthebiologically functionalHTLV-Iprotease gene and that the active form of the HTLV-I protease molecule
processesthegag precursorpolyprotein in trans(Fig. 5).
Asignificant structural differencebetween HTLV-I prote-ase and other retroviral proteases is the length ofthe pro
ORF. The 3' region of the putative HTLV-I protease,
extending from the second conserved aminoacid stretch to the carboxy-terminal residue, is more than 46amino acids
longer than the analogous region ofother members ofthe HTLVfamily(26). The datapresentedin this paper demon-stratethatthisregion is dispensable for
proteolytic
activity oftheHTLV-Iprotease,despite
thehighlevelofaminoacid homology found in this region among the members ofthe HTLVfamily (Fig. 3).Thebiological function ofthisregion
remains unclear.
A survey of the amino acid sequence of the HTLV-I protease revealed the presence of thetwostrictly conserved regionscommon totheretroviralproteases. Theuniquecore sequence, D-T-G,
constituting
one of the conserved se-quences, was identical to that ofaspartyl protease, which has been predicted as the catalytic center for proteaseactivity (30, 44). We introduced a mutation in this
aspartic
acid,atposition64in HTLV-I protease, and found that the
mutation completely abolished cleavage. Independent evi-dence suggesting that the retroviral protease is an aspartyl protease has recently been obtained enzymatically with aspartyl protease inhibitors (16). Furthermore, we have
foundthat oneAsp-to-Gly changeatamino acid position125 inHTLV-Iproteasealsoabolished the processing of the gag precursor. The aspartic acid at amino acid 125 is located
withina shortaminosequence L-V-D-T, which is not found among theproteasesof animaltypeB, C,or Dretrovirusor
lentivirus or among cellular aspartyl proteases. Interest-ingly, thissequence, L-V-D-T, or its homolog isconserved onlyinthe proteaseoftheHTLV,family including HTLV-I,
HTLV-II, and bovine leukemia virus (34, 49). Molecular
aspects of HTLV-I protease biosynthesis have not been elucidated so far. The coding frame of the pro gene is different fromthatofthe gag gene,andnomethioninecodon was found at the 5' terminal of the pro ORF. Therefore,
either mRNA processing or translational frame shifting would be required to produce functional protease. Correct processing ofthe gag precursorpolyprotein in thevaccinia
virus expression vector system strongly suggests that a
frameshiftmechanismwould beinvolvedin protease synthe-sis, because this system does not support thesplicingevent of mRNA. Infact, characteristic nucleotide sequenceswere found in the
overlapping
region between the gag and pro genes, which contains aguanine-plus-cytosine-rich
hairpin
structureand six consecutive
adenine
residues. Bothchar-acteristic sequences havebeen
thought
tobeimportant
forribosomal frame
shifting
on mRNA in procaryotes and eucaryotes (1, 5, 19). We directedsite-specific
mutations into the six consecutive adenine residues to create a new sequence, AATATT. Theprocessing
defect ofthis mutantmay becausedbythe
blocking
of ribosomal frameshifting
responsiblefor
synthesis
of the protease,suggesting
thatthe gag-pro fusionprotein
wouldbetranslatedas aprecursor of a protease via ribosomal frameshifting.
Our dataare com-patiblewith thoseformousemammary tumorvirus(11, 15).
Thegag-pro precursor
polyprotein
isexpected
to be 76 kilodaltons in size.However,
a 76Kprotein
has not beenVOL.62, 1988 3725
-I_A
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.109.519.72.263.2]3726 NAM ET AL.
1 2 3 4 5
5 3 K
-- 2 4 K
:-19K 1- 1;o.
FIG. 9. Identification of 76K gag-protease fusion protein. (A) Schematic representation of the mutant plasmids which express gag-proteasefusion proteins. Symbols: 0, 7.5K genepromoterof vaccinia virus; , twoconserved amino acid stretches between the
retroviral proteases and the cellular enzyme group of aspartyl
protease;
0,
short amino acid residues conserved only amongHTLV-family retroviral proteases; +or, in vitro-mutagenized amino acidresidues described in Fig. 6A; ,junction betweenthe
gag and protease-coding sequences in fusion proteins; ,
pUC DNA sequence. (B) Analysis of gag-protease fusion
pro-teins expressed from the mutants by Western blotting. Proteins extracted from transfected cell with p7.5gagfsl9Aspl (lane 1), p7.5gagfsl9Asp1I (lane 2), p7.5gagfsl9AspIAOE (lane 3), and
p7.5gagfsl9AspIIAKp (lane 4) were separated on an SDS-12% polyacrylamidegel, transferredtoanitrocellulosefilter, andprobed
with mixed anti-gag monoclonal antibodies of GIN7, NORI, and FR45.MT-2celllysatewasusedasthe markerof authenticmature
gagproteins (lane 5).
identified inthechronicallyinfected cell line. In vitro
trans-lation system programmed withvirion-associated RNA al-lowed the detection of trace amounts ofa 76K protein by immunoprecipitation withserumfromanadultT-cell leuke-miapatient (18).Consideringthatthevirion-associated RNA directsonly thesynthesisofgaggene-related products in the invitrotranslationsystem,wepresumethat the 76Kprotein observed in invitro-translatedproducts is thegag-profusion
protein. Tworeasons for failuretodetect this precursor are
conceivable: rapid self-cleavage of the frameshift productor
the inefficiency of ribosomal frame shiftingfor synthesis of gag-pro precursorpolyproteins, orboth.
To examine the action ofprotease on the 76K gag-pro
precursorpolyprotein, we introducedaninsertion mutation
to align both frames of thegag andprogenes, producinga
detectable amount of thisprecursor. Although this gag-pro
fusion protein is not exactly the same as the precursor
protein produced by ribosomalframe shift, itshouldbevery
similar(Fig. 9). Correct processing ofthe 76K protein could beobservedonly when it retainedanactiveproteasedomain (Fig. 8). This result stronglysuggests that HTLV-Iprotease
is capable oftriggering the first cleavage eventfor its own releasefromthegag-pro precursor,
probably
in acis-acting
manner.
Autocatalytic cleavage ofviralproteasehasbeen
reported
inRNA viruses such as
picornaviruses (7, 28, 40)
butnotinretroviruses (47, 48). Accordingtoa recent
study, however,
human immunodeficiency virus protease
expressed
inSac-charomyces cerevisiaeandE. coliexhibited
autoprocessing
ofthe gagprecursormodel(4,
21).
Our results with HTLV-I protease also support the autocatalytic cleavage model.Thus, it appears likely that self-cleavage of constituent proteinsmightbeacommonphenomenonin thebroadrange
ofvirusspecies including retroviruses. However,wecannot excludethe
possibility
that acellularprotease activates the inactiveprecursor.
Deduced amino acid sequencealignment ofgag-profusion protein among retroviruses implies that protease
might
beinvolvedin virusreplicationor
integration
into host genome(17). We are now in the processof
establishing
an invitroenzyme assay system
involving
purified
protease and thepurified gag precursor product of HTLV-I. The system
should provideus with the
precise
enzymatic
properties
ofHTLV-Iprotease. Suchknowledge will facilitate the
devel-opmentof effective antiviral
therapeutic
agents. ACKNOWLEDGMENTSWethankY.ItoandM.Maki formakinghelpfulsuggestionsand K. Igarashi, Takeda Chemical Industries, Ltd., for synthesizing oligonucleotides.
This work was supported by grants-in-aid from the Ministry of Education,Science, and Culture of Japan.
ADDENDUM
Jacks et al. (14) have recently shown by amino acid
sequencing
and in vitromutagenesis
that the consecutive uridine residues located upstream of theputative
guanine-plus-cytosine-rich
hairpin structure are involved in ribo-somal frame shifting for thesynthesis
ofgag-pol
fusion protein inhumanimmunodeficiency virustype I.Their result isconsistent with ourdataforlocating the candidate site of ribosomalframeshift.In addition, several groups have recently reported inde-pendent results which support an
enzymatic
role for theconserved Asp in the D-T-G sequence in retroviral prote-ases, homologous tothecatalyticcenterin cellular
aspartyl
proteases. Changes ofthis Asp to Ala in the human immu-nodeficiency virus protease (25) and to Ile in the avian sarcoma-leukosis virus protease (20) have been shown to
abolish cleavage ofthe human immunodeficiency virus and avian sarcoma-leukosis virusprotease-containing viral pre-cursor
proteins,
respectively. Thus, any one of three changes at the site (Asp to Gly, Ala, or Ile) appears todestroy protease activity in three differentviral proteases. LITERATURECITED
1. Atkins, F., R. F. Gesteland, B. R. Reid, and C. W. Anderson. 1979. Normal tRNAspromoteribosomalframeshifting.Cell 18: 1119-1131.
2. Birnboim, H. C., and J. Doly. 1979. A rapidalkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res.7:1513-1523.
3. Cochran, M. A., C. Puckett, and B. Moss. 1985. In vitro
mutagenesis of the promoterregion foravaccinia virusgene: evidence fortandemearly and late regulatory signals.J. Virol. 54:30-37.
4. Debouck,C., J. G. Gorniak, J. E. Strickler, T. D. Meek, B. W. J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.612.63.292.76.343.2]PROCESSING OF HTLV-I gag POLYPROTEIN BY VIRAL PROTEASE Metcalf, and M. Rosenberg. 1987. Human immunodeficiency
virus protease expressed inEscherichia coliexhibits autopro-cessing and specific maturation of the gag precursor. Proc. Natl. Acad. Sci. USA 84:8903-8906.
5. Fox, T. D., and B. Weiss-Brummer. 1980. Leaky +1 and -1 frameshift mutations at the same site in a yeast mitochondrial gene. Nature(London)288:60-63.
6. Graham, F. L., and A. J.vander Eb. 1973. A new technique for the assay of infectivity of humanadenovirus DNA. Virology 52: 456-467.
7. Hanecak, R., B. L.Semler, H.Ariga, C. W.Anderson,and E. Wimmer. 1984. Expression of a cloned gene segment of polio-virus in E. coli: evidence forautocatalytic production ofthe viralproteinase. Cell 37:1063-1073.
8. Hattori, S., T. Kiyokawa, K. Inakawa, F. Shimizu, E. Hashi-mura, M. Seiki, andM.Yoshida. 1984. Identification of gag and env gene products of human T-cell leukemia virus (HTLV). Virology 136:338-347.
9. Hinuma, Y., K. Nagata, M.Hanaoka, M.Nakai,T. Matsumoto, K. Kinoshita, S.Shirakawa, and I. Miyoshi. 1981. AdultT-cell leukemia: antigen in an ATL cell line anddetection of antibodies tothe antigen in human sera. Proc. Natl. Acad. Sci. USA 78: 6476-6480.
10. Hiramatsu, K., J. Nishida, A. Naito, and H. Yoshikura. 1987. Molecular cloning of the closed circular provirus of human T cell leukaemia virus type I: a new open reading frame in the gag-pol region. J. Gen. Virol. 68:213-218.
11. Hizi, A., L. E.Henderson, T.D. Copeland, R. C. Sowder, C. V. Hixson, and S.Oroszlan. 1987. Characterization of mouse mam-mary tumor virus gag-pro gene products and the ribosomal frameshift site by protein sequencing. Proc. Natl. Acad. Sci.
USA 84:7041-7045.
12. lino, T., K. Takeuchi, S. H. Nam, H. Siomi, H. Sabe, N. Kobayashi, and M. Hatanaka. 1986. Structural analysis of p28 adult T-cell leukaemia-associated antigen. J. Gen. Virol. 67: 1373-1379.
13. Inoue, J., T. Watanabe, M. Sato, A. Oda, K. Toyoshima, M. Yoshida, and M. Seiki. 1986. Nucleotide sequence of the prote-ase-coding region in an infectious DNA of simian retrovirus (STLV) ofHTLV-I family. Virology 150:187-195.
14. Jacks, T., M. D. Power, F. R. Masiarz, P. A. Luciw, P.I. Barr, and H. E. Varmus. 1988. Characterization of ribosomal frame-shifting in HIV-1gag-pol expression. Nature (London) 331:280-283.
15. Jacks, T., K.Townsley, H. E. Varmus, and J. Majors. 1987. Two efficientribosomal frameshifting events are required for synthe-sis of mouse mammary tumor virus gag-related polyproteins. Proc. Natl. Acad. Sci. USA84:4298-4302.
16. Katoh, I., T. Yasunaga, Y. Ikawa, and Y. Yoshinaka. 1987. Inhibitionofretroviral protease activity by an aspartyl protein-ase inhibitor. Nature (London)329:654-656.
17. Katoh, I., Y.Yoshinaka, A. Rein, M. Shibuya, T. Okada, andS. Oroszlan. 1985. Murine leukemia virus maturation: protease region required forconversion from "immature" to "mature" core form andfor virusinfectivity. Virology 145:280-292. 18. Kobayashi, N., N. Yamamoto, Y. Koyanagi, J. Schneider, G.
Hunsmann, and M. Hatanaka. 1984. Translation of HTLV (human T-cellleukemia virus)RNAinanuclease-treatedrabbit reticulocyte system. EMBO J. 3:321-325.
19. Kohno, T., and J. R. Roth. 1978. A Salmonella frameshift suppressor that acts at runs of A residues in the messenger RNA. J. Mol. Biol. 126:37-52.
20. Kotler, M. R., A. Katz, and A. M. Skalka. 1988. Activity of avian retroviral protease expressedinEscherichiacoli.J.Virol. 62:2696-2700.
21. Kramer, R. A., M. D. Schaber, A.M. Skalka, K. Ganguly, F. Wong-Staal, and E. P. Reddy. 1986. HTLVIII gag protein is processed in yeast cells bytheviruspol protease. Science231: 1580-1584.
22. Laemmli, U. K. 1970. Cleavage of structuralproteinsduringthe assembly of the head of bacteriophage T4. Nature (London) 227:680-685.
23. Maniatis, T., E. F. Fritsch, andJ. Sambrook. 1982. Molecular
cloning: a laboratory manual. Cold Spring Harbor Laboratory, ColdSpringHarbor,N.Y.
24. Mori,K., H.Sabe, H. Siomi, T. lino, A. Tanaka, K. Takeuchi, K.Hirayoshi, and M. Hatanaka. 1987. Expression of a provirus ofhuman T cellleukaemiavirustypeIbyDNAtransfection. J. Gen. Virol. 68:499-506.
25. Mous, J., E. P. Heimer, and S. T. J. LeGrice. 1988. Processing protease and reverse transcriptase from human immunodefi-ciency virustypeI polyprotein in Escherichiacoli.J. Virol. 62: 1433-1436.
26. Nam, S. H., and M. Hatanaka. 1986. Identification of a protease gene ofhumanT-cell leukemia virus type I (HTLV-I) and its structuralcomparison. Biochem. Biophys. Res. Commun. 139* 129-135.
27. Oroszlan,S., and T. D.Copeland. 1985. Primary structureand processing of gag and env gene products of human T-cell leukemia virus HTLV-ICR HTLV-IATK. Curr. Top. Microbiol. Immunol. 115:221-223.
28. Palmenberg, A. C., and R. R. Rueckert. 1982. Evidence for intramolecular self-cleavage of picornaviral replicase precur-sors.J. Virol. 41:244-249.
29. Pearl, L. H., and W. R. Taylor. 1987. Sequence specificity of retroviralproteases. Nature (London)328:482.
30. Pearl,L.H., and W. R. Taylor. 1987.A structuralmodelforthe retroviralproteases. Nature (London) 329:351-354.
31. Poiesz, B. J.,F.Ruscetti, A. F. Gazdar, P. A. Bunn, J. D.Minna, and R. C. Gallo. 1980. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patientwith cutaneousT-cell lymphoma.Proc.Natl. Acad.Sci. USA77:7415-7419.
32. Poiesz, B. J., F. W. Ruscetti, M. S. Reitz, V. S. Kalyanaraman, and R. C. Gallo. 1981. Isolation of a new type C retrovirus (HTLV) in primary uncultured cells of a patient with Sezary T-cellleukemia. Nature (London) 294:268-271.
33. Ratner, L., S. F. Josephs, B. Starcich, B. Hahn, G. M. Shaw, R. C. Gallo, and F. Wong-Staal. 1985. Nucleotide sequence analysis ofa variant human T-cell leukemia virus (HTLV-Ib) provirus with a deletion in pX-I. J. Virol. 54:781-790. 34. Sagata,N., T.Yasunaga, J.Tsuzuku-Kawamura, K. Ohishi, Y.
Ogawa, and Y. Ikawa. 1985. Complete nucleotide sequence of thegenome ofbovineleukemia virus: its evolutionary relation-shiptoother retroviruses. Proc. Natl. Acad. Sci. USA 82:677-681.
35. Sanger, F., S. Nicklen, andA. R.Coulson. 1977. DNA sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467.
36. Schwartz, D. E., R. Tizard, and W. Gilbert. 1983. Nucleotide sequence ofRous sarcoma virus. Cell32:853-869.
37. Seiki, M., S. Hattori, Y. Hirayama, and M. Yoshida. 1983. Human adult T-cell leukemia virus: complete nucleotide se-quence of the provirusgenomeintegrated in leukemia cell DNA. Proc. Natl. Acad. Sci. USA 80:3618-3622.
38. Shida, H. 1986. Nucleotide sequence of the vaccinia virus hemagglutinin gene.Virology 150:451-462.
39. Shida, H., T. Tochikura, T. Sato, T. Konno, K. Hirayoshi, M. Seki, Y.Ito, M. Hatanaka, Y. Hinuma, M. Sugimoto,F. Taka-hashi-Nishimaki, T. Maruyama, K. Miki, K.Suzuki,M.Morita, H.Sashiyama,and M. Hayami. 1987. Effect of the recombinant vacciniavirus that express HTLV-I envelope geneon HTLV-I infection. EMBO J. 6:3379-3384.
40. Strebel, K., and E. Beck. 1986. A second protease of foot-and-mouth disease virus. J. Virol. 58:893-899.
41. Tanaka, Y., Y. Koyanagi, T. Chosa, N. Yamamoto, and Y. Hinuma. 1983. Monoclonal antibodies reactive with both p28 and p19 of adult T-cell leukemia virus-specific polypeptides. Gann 74:327-330.
42. Tanaka, Y., B. Lee, T. Inoi, H. Tozawa, N.Yamamoto, and Y. Hinuma. 1986.Antigensrelatedtothreecoreproteinsof HTLV-I (p24, p19 and p15) and their intracellular localizations, as
defined by monoclonal antibodies. Int. J. Cancer 37:35-42. 43. Taylor, J. W., J. Ott, and F. Eckstein. 1985. The rapid
genera-tion of oligonucleotide-directed mutations at high frequency using phosphorothioate-modified DNA. Nucleic Acids Res. 13:
VOL.62, 1988 3727
on November 10, 2019 by guest
http://jvi.asm.org/
3728 NAM ET AL. 8765-8785.
44. Toh, H.,R.Kikuno,H.Hayashida,T.Miyata,W.Kugimiya,S.
Inoue,S. Yuki, and K. Saigo.1985. Close structural resemblance between putative polymerase of a Drosophila transposable
genetic element 17.6 andpolgeneproduct of Moloney murine
leukaemia virus. EMBO J. 4:1267-1272.
45. Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels tonitrocellulose sheets:procedure andsomeapplications. Proc. Natl. Acad. Sci. USA76:4350-4354.
46. Venkatesan, S., B. M. Baroudy,and B.Moss. 1981. Distinctive
nucleotide sequences adjacenttomultiple initiation and
termi-nation siteof early vaccinia virusgene.Cell125:805-813.
47. Vogt, V. M., R.Eisenman, and H. Diggelmann. 1975. Generation ofavianmyeloblastosis virus structuralproteins by proteolytic cleavage ofaprecursorpolypeptide. J. Mol. Biol. 96:471-493.
48. vonder Helm, K.1977. Cleavage of Roussarcomaviral
polypep-tideprecursorintointernal structural proteins in vitro involves viral proteinp15. Proc. Natl. Acad. Sci. USA 74:911-915.
49. Yasunaga, T., N. Sagata, and Y. Ikawa. 1986. Protease gene
structure and env gene variability of the AIDS virus. FEBS
Lett. 199:145-150.
50. Yoshida, M., I. Miyoshi, and Y. Hinuma. 1982. Isolation and
characterization of retrovirus from cell lines of human adult T-cellleukemia and its implication in the disease. Proc. Natl. Acad. Sci. USA 79:2031-2035.
51. Yoshinaka, Y.,I.Katoh,T. D. Copeland, and S.Oroszlan. 1985.
Translational readthrough ofanambertermination codon during
synthesis of feline leukemia virusprotease.J. Virol. 55:870-873. 52. Yoshinaka,Y.,I.Katoh,T. D.Copeland, and S.Oroszlan. 1985.
Murineleukemia virusproteaseisencoded by thegag-polgene
and issynthesized through suppression ofanambertermination
codon. Proc. Natl. Acad. Sci. USA 82:1618-1622.
53. Yoshinaka, Y., I.Katoh, T. D. Copeland, G. W.Smythers, and
S.Oroszlan. 1986. Bovineleukemia virusprotease:purification, chemical analysis and in vitro processing of gag precursor
polyproteins. J. Virol. 57:826-832.
J. VIROL.