0022-538X/92/031688-07$02.00/0
Copyright© 1992, AmericanSocietyforMicrobiology
Binding of
a
Host Cell Nuclear Protein
to
the
Stem
Region of
Human Immunodeficiency
Virus
Type
1
trans-Activation-Responsive RNA
MATTHEW P. ROUNSEVILLE' ANDAJIT
KUMAR'
2*Graduate Program in Genetics' and Department of Biochemistry and MolecularBiology,2 theGeorge Washington University Medical Center, Washington, D.C. 20037
Received3September 1991/Accepted7December1991
Humanimmunodeficiency virustype1(HIV-1) transcription is regulated by both viral and host cell factors.
Although the viral trans-activator protein, Tat, and its cis-responsive element, trans-activation-responsive (TAR) RNA, have been identified and characterized, the mechanism ofHIV-1transcriptionalregulation has notbeen satisfactorily described. Whereas Tat isnecessarytoactivatetranscription,additionalfactors, derived from the host cell,areimportant inregulating HIV-1transcription. Toidentify such host cell-specific factors,
weusedanRNase protection mobility shiftassayand UVcross-linking todetecta 140-kDa HeLa cell nuclear protein that bindsspecificallytoTARRNA.By extensive mutationalanalysis,wedetermined that thebinding of thisprotein is dependentonboth thesequenceandthe structureof theTAR RNA stemregion.Othergroups
have shown that the production of prematurely terminated transcripts from the HIV-1 promoter is also dependentonthesequenceandstructureofthe TAR RNAstem.Thiscorrelation withourresults suggeststhat the TAR RNAstem-binding protein is involvedintheproduction of prematurelyterminatedtranscriptsfrom the HIV-1promoterand intheregulation of HIV-1 geneexpression.
Human immunodeficiency virustype 1(HIV-1) transcrip-tion is regulated by viral and cellular factors, whichact on
viralcis-responsive elementstoeither induceorrepressviral
gene expression (for reviews, see references 4 and 18). The HIV-1 trans-activator protein, Tat, interacts with its cis-responsive element, trans-activation-responsive (TAR) RNA, to increase greatly the synthesis of full-length viral transcripts. TAR forms a stable stem-loop structure and is located at nucleotide positions +1 to +57 within the 5' untranslated region of all viral mRNAs (3, 14) (Fig. 1). Although Tat binds tothepyrimidine bulge and the immedi-ateflanking base-paired sequencesof TAR RNA invitro (6, 19, 23), this interaction isnotsufficienttoactivate transcrip-tion in vivo (6, 19). For example, Tat bindsto TARRNAs withmutations in the loop; however, these mutations donot supportTat-mediated transactivation in vivo. This suggest that host cell factors are involved in regulating the HIV promoter. Indeed, Barryetal. (1) have shown that levels of HIV-1transcriptional activityvaryupto1,000-fold between cell types. Further, studies of rodent-human cell hybrids demonstrate thatwhereas certainrodent cells, suchasCHO cells, either donot supportTat-mediated transactivationor do so poorly, Tat-mediated trans activation is increased markedly in rodent-human hybrid cells which retain human chromosome 12(9, 16). Additionally, Marciniaketal. have identifieda68-kDa HeLa cell nuclear protein which bindsto
theloop of TAR and increases transactivation in vitro (12, 13). These observations demonstrate that host cell factors playanimportant role in regulating HIV transcription.
To identify such host cell-specific factors, we used an
RNase protection mobility shift assayand UV cross-linking
to detect a 140-kDa HeLa cell nuclear protein that binds
specifically to TAR RNA. Through extensive mutational analysis, we determined that the binding of this protein is
*Correspondingauthor.
dependent on both the sequence and the structure of the TAR RNA stem region. Interestingly, other groups have shown that the production of prematurely terminated tran-scripts from the HIV-1 promoter is dependent on the se-quenceandstructureof the TAR RNAstem(11, 17, 20, 21). ThiscorrelationwithourresultssuggeststhattheTAR RNA stem-binding protein(SBP) is involved in the productionof prematurely terminated transcripts and in the regulationof HIV-1geneexpression.
(This research wasconductedby M.P.R. in partial fulfill-ment of the requirements for a Ph.D. in genetics from the GeorgeWashington University, Washington, D.C., 1992.)
MATERIALS ANDMETHODS
Plasmidconstructs and invitro transcription. An in vitro transcription-cassette vector, pT7pUC19, was constructed
by cloning the bacteriophage T7 RNA polymerasepromoter
(5'-TAATACGACTCACTATA-3') into theEcoRI-KpnI site ofpUC19 (Bethesda Research Laboratories, Inc., Gaithers-burg, Md.). Synthetic TAR and TARmutant oligodeoxynu-cleotides(synthesizedon anApplied Biosystems 380B DNA
synthesizer) were then cloned into the KpnI and HindlIl
sitesofpT7pUC19. The plasmids werelinearizedatHindlIl and transcribed in vitro with [a-32P]CTP or [a-32P]UTP (>400 Ci/mmol) (Amersham, Arlington Heights, Ill.)
essen-tially as described by the supplier of the transcription kit (Promega, Madison, Wis.). Aftertreatmentwith RNase-free DNase(1U/4LgofDNA, 37°C, 15min), the transcriptswere purifiedonan 8%bisacrylamide-7 Mureagel, eluted in 0.5
M ammonium acetate-1 mM EDTA-0.1% sodium dodecyl sulfate(SDS), extractedonceinphenol-chloroform andonce inchloroform, ethanol precipitated, and resuspendedto 105 cpm/lulinwater.Thesecondarystructuresand free energies of the mutant TAR RNAs were predicted with an RNA-foldingcomputerprogram(26).
RNaseprotection mobility shiftassay. TheRNase
protec-1688
on November 10, 2019 by guest
http://jvi.asm.org/
30
BulgooCUGZGCC
Laop
20 G
I/AG-
CUC GC 40
IA-U
U-AU-A
CuG=A 10 G . U
U-A-50 cG
lU
- Gc-G IU-A C ,A
FIG. 1. Secondary structure of HIV-1 TARRNA positions +1 through +57. TAR RNA has been divided into sections as follows: fourstemregions (regionI,bases 5 to 9 and 50 to 54; regionIl,bases 10to 15and44to49; region III, bases 17 to 21 and 39 to 43; and regionIV, bases 25 to 28 and 35to38),a3-base pyrimidinebulge (U22C23U24),asix-member loop(CUGGGA), and unpaired nucleo-tides(C4 and
Al6).
tion mobility shift assay is a modification of one used to demonstrate sequence-specific binding of the HIV-1 Rev proteintothe Rev-responsive elementRNA (25). The reac-tion mixtureconsisted of 50%(vol/vol)nuclear extract (5 to 10 mg/ml in buffer D, which consisted of 20 mM HEPES [N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid] [pH 7.9], 100 mM KCl, 20% [vol/vol] glycerol, 0.5 mM phenyl-methylsulfonyl fluoride, and 0.5 mMdithiothreitol [5]), 1.5 mMMgCl2, and105 cpmofradiolabelled transcript,in a total volume of10
p.J.
Thereaction mixturewas incubated for 15 min at room temperature, and then 1 ,ul of a solution of RNasesA(1mg/ml) andT1 (5,000 U/ml) was added and the incubation was continued for 15 min. One microliter of loading dye(97%
glycerol, 0.1% xylene cyanol, 0.1% bro-mophenol blue) was added, and the reaction mixture was loaded on a 4% native polyacrylamide gel(acrylamide-bisacrylamide
[80:1]-50
mMTris-50mMglycine [unadjustedpH - 8.8]) andrunat100 Vfor5h oruntilthexylene cyanol
had migrated approximately 12cm. Thegel was then dried and exposed to X-ray film overnight at -70°C with an intensifying screen. Competitionassays were performedby incubating the nuclearextractwithnonradiolabelled compet-itor RNA for 10 min prior to adding the 32P-labelled TAR RNA.
UVcross-linking. The
binding
reactionswereperformed
asdescribed above. After the RNase treatment, the samples were transferred to ice and irradiated for 30 min with shortwave UV
light
at8 mW/cm2(measured
at thesource)
fromadistance of 5cm.Anequal volume of2x SDSsample buffer was then added, and the samples were placed in a100°C water bath for 5 min and separated on an SDS-6%
polyacrylamide gel.
Thedye frontwasallowedtomigrate
offthegelto remove
32P-labelled degradation products.
Thegel
wasthen fixedin 40%methanol-10% aceticacid, dried,
andexposed
toX-ray filmat -70°Cwithanintensifying
screen.q,
GI)
k. 4zp
z Cli Ol ,.& 13,
0
00- 0
RNase
digestion
products
[image:2.612.116.262.77.285.2]2 3 4 5
FIG. 2. RNaseprotection mobility shift assay.An RNase-resis-tant RNA-protein complex was formed when
32P-labelled
TAR RNAwasincubated with HeLa nuclear extract (NE) (arrow, lane 2) but was not formed in the absence of nuclear extract (lane 1). Complexformationwasabolished when RNasewasaddedpriorto theaddition of nuclear extract (lane 3), when0.1% SDSwaspresent (lane 4), and when the nuclear extract was treated with 2 ,ug of proteinaseK(lane 5).RESULTS
A HeLa
cell
nuclear protein binds to TAR RNA. To investigate whether a HeLa cell nuclear protein forms aspecific
complex with TARRNA, radiolabelled TAR RNAwasincubated withHeLa nuclear extract, and then nonspe-cifically bound RNA was digested with RNases A andT1. Theresulting complexwasresolvedon ahigh-ionic-strength, low-cross-linked, native polyacrylamide gel. Adistinct band of alteredelectrophoretic mobilitywasobservedwhen TAR RNA wasincubated withHeLanuclearextract(Fig. 2,lane 2). This complexwas notformedwhentheRNase wasadded priorto theaddition ofnuclear extract(Fig. 2,lane 3).This control shows that the protected RNA (lane 2) is not an inherently RNase-resistant fragment bound nonspecifically to protein or hybridized to nucleic acid in the nuclear extract.Thecomplexwasalso notformedin thepresenceof 0.1% SDS
(Fig.
2, lane4) orwhen the extractwas preincu-bated withproteinase
K(Fig.
2, lane 5). The absence of complex formation under these conditions demonstratesthe RNA-proteinnatureof thecomplex.
ThisTAR RNAprotein
bindingactivitywasalso detected in HUT-78 nuclearextract(datanotshown).
TARRNAdeletionanalysis.Todeterminethesequences of TAR RNA essential to the formation of this
RNA-protein
complex,severaldeletionmutantsofTARwereconstructed and tested in thegel
mobility
shift assay(Fig.
3 showsthe sequences andFig.
4 shows thepredicted secondary
struc-turesand stabilitiesofall TAR RNAmutants).
InTM12,
thestem
regions
III(bases
17to21and 39to43)
and IV(bases
25 to28 and 35to
38)
andthepyrimidine bulge
(U22C23U24)
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.379.507.87.331.2]1 10 20 30 40 50 WTTAR GGUCUCUCUGGUUAGACCAGAUCUGAGCCUGGGAGCUCUCUGGCUAACUAGGGAACC
TM12 GGUCUCUCUGGUUAG ---CCUGGGA---CUAACUAGGGAACC
TM27 GG---CCAGAUCUGAGCCUGGGAGCUCUCUGG---CC TM30 GGUCUCUCUGGUUAGACCAGAUCUGAGCCU---AGCUCUCUGGCUAACUAGGGAACC
TM29 GGUCUCUCUGGUUAGACCAGA---GAGCCUGGGAGCUCUCUGGCUAACUAGGGAACC
TM31 GGUCUCUCUGGUUAGACCAGAUCU---CUGGCUAACUAGGGAACC
TM40
---GUUAGACCAGAUCU---CUGGCUAAC---TM38
GGUCUCUCUGGUUAGACCAGAUCUGAGCCUGGGAGCUCUCUGGCUA---TM18 G9CAfi99&q9hMAaMUC
TM22 GGUC&QMAGGUUAGACCAGAUCUGAGCCUGGGAGCUCUCUGGCUAACUUUfACC
TM23 GGUCUCUCU=^CCAGAUCUGAGCCUGGGAGCUCUCUGGgAMJAGGGAACC
TM24 GGUCUCUCUGGWAGA99CUCUGAGCCUGGGAGCUCAQACCUAACUAGGGAACC
TM25 GGUCUCUCUGGUUAGACCAGAUCUCJCUGGGAC9UCUGGCUAACUAGGGAACC
TM26 GGUCUCUCUQGUAGACCAGAGAGAGCgACCGCUCUCUGGCUAACUAGGGAACC
TM37 GGUCUCUCUGGUUAGACIAGAUCUGAGCCUGGGAGCUCUCUGGCUAACUAGGGAACC
TM39 GGUCUCUCUGGUUAG-CCAGAUCUGAGCCUGGGAGCUCUCUGGCUAACUAGGGAACC
FIG. 3. Sequence ofwild-type(WT) TARRNAandmutantTAR RNAswhich have beencloned into the in vitrotranscriptionvector
pT7pUC19. Deletions are indicated by dashes; base substitutions areboldface and underlined.
have beendeleted, whereasthe lower stem regionsI
(bases
5 to 9and 50to54) andII(bases
10 to 15and44to49) and the six-member loop (CUGGGA) have been retained. In asecond deletionmutant,TM27, the lowerstem
regions
Iand II, the unpaired residues C4 and A16, and the basepair
U3 * A55havebeendeleted,whereastheupperportion ofthe molecule (stemregions IIIandIV, thepyrimidine bulge,
and theloop) andthefirsttwoG C basepairs ofthestemhave beenleft intact(Fig.
4). Nuclearprotein
binding
tothe TM12 transcript was abolished (Fig. 5, lane 3).Thus,
the stemregions IandII andtheloop sequences arenotsufficientto support the RNA-protein complex formation. This result suggests thatregions within theupper
portion
ofTAR RNA (the unpaired adenine atposition +16, stemregion III,
the UCUbulge, and stemregion
IV) are necessaryforprotein
recognition. Proteinbinding to the transcript ofTM27 was detectable yet substantially reduced (Fig. 5, lane 4). This demonstrates that while TM27 may contain the minimum sequencesnecessaryforproteinrecognition, the lowerstem regions (deleted in this mutant) play an important role in stabilizingtheRNA-protein interaction.The TAR RNA pyrimidine bulge, stem IV, and the loop areessential forTat transactivation(19-21). To test whether these regions are essential to the binding of the host cell nuclear protein, we constructed three TAR mutants with deletions in these
regions.
The three Gresidues of the loop (G31G32G33) were deleted in TM30, the pyrimidine bulge (U22C23U24) was deleted in TM29, and both the loop and stem region IV(sequences 25 to 38) were deleted in TM31 (Fig. 4). Neither the 3-basedeletion in the loop (TM30)nor the more extensive deletion(TM31) had an effect on protein binding (Fig.5,
lanes 5 and 7,respectively). Protein binding to the bulge deletion mutant (TM29), however, was mark-edly reduced (Fig. 5, lane 6; an additional, faster-migrating RNA-protein complex wasformed with the TM29 mutant, presumably because oftheextended9-bp stem between A16 and the loop). These deletion studies establish that stem region IV and the loop are not essential for the TAR RNA-protein complex formation and indicate that thepy-rimidine bulge
isimportant.
Twolower-stemdeletionmutants werecreatedtoexamine the contribution of this region to the TAR RNA-protein interaction. Inthe mutantTM40,theloopandstem regions Iand IV have been deleted(compare TM31,in whichjustthe loop and stemregion IV have beendeleted; Fig. 4),and in TM38 bases 47 to 57 have been deleted
(Fig. 4).
The TARRNA-binding protein
did notformacomplex
with either ofthese mutant TAR RNAs (Fig. 5, lanes 8 and 9). This experiment confirms that a structured
base-paired
stem in the lowerregionof TAR RNA isnecessarytosupport TAR RNAproteinbinding activity.
TAR RNA primary sequence analysis. We next
analyzed
theeffects ofTAR RNAprimarysequence mutationsontheTAR
RNA-protein
interaction. Several compensatorymuta-tions were designed to alter theprimary sequence of TAR and yetmaintainasfaras
possible
thesecondary
structureofthe
wild-type
molecule. InTM18,
the sequences of stemregions I, II, III, andIVhavebeenswitched
(for
example,
aG C has become a C G), whereas the
non-base-paired
nucleotides(thecytosine
residueatposition +4,
theadenine residue at position +16, and thebulge
andloop)
have notbeen altered. The mutations in TM18 have the
predicted
effects ofcollapsing
thepyrimidine bulge,
ofleaving only
theU22
unpaired,
and ofaltering
the structureof theloop
andstemregionIV
(Fig.
4).The TM18transcript
was notbound by the nuclearprotein (Fig.
5, lane10).
This result shows that the primary sequence of the stem isimportant
for proteinrecognition
and demonstrates that the presence of thesinglybulged nucleotides(C4
andA16) isnotsufficientto supportprotein binding.We constructed several base
pair-switching
mutants(TM22,
region
I; TM23,region II;
TM24,region III;
and TM25, region IV)(Fig.
4) to determinespecifically
which region ofthe TM18 RNA molecule isresponsible
for dis-missingprotein
binding
activity.
Theprimary
sequence alterationsin stemregions
I(TM22)
and II(TM23)
greatly
reducedprotein
binding
activity (Fig. 5,
lanes11and12)
and therebydemonstratethat theprimary
sequenceof the lower two stemregions isimportant
forthe stablebinding
of the hostcell factor. Theprimary
sequence switch instemregion
III (TM24) has thepredicted
effects ofreducing
the5-bp
stemIII toa4-bp
stemandofaltering
thecomposition
of the bulgefrom UCUto UUC(Fig.
5). Proteinbinding
toTM24 RNA waslessefficientthan to thewild-type
TAR RNA(Fig.
5, lane13). The implication fromthisexperiment is that the primary sequenceofstem regionIIIandthecomposition ofthe
pyrimidine
bulge contribute tostable TARRNA-protein
binding. The base
pair
switches in stem IV (TM25) altersubstantially
thepredicted
composition
andstructure ofthebulge, stem region IV, and the loop
(Fig.
4). For example, thebulge is reducedto asingle residue (U22), thestemregion
IVisextendedintoaninterrupted
5-bpstem, and theloopis curtailed to five nucleotides insteadofthe usual six. Surpris-ingly, TM25RNAbound tothe nuclear proteinwith alevelof
activity
somewhat higher than that ofwild-type
TARRNA
(Fig.
5, lane 14). The mutational analysis describedabove demonstrates that the
primary
sequence ofthe stemregionsI, II,and IIIplaysanimportantrole in thebindingof thishost cell factor toTAR RNA.
To extendthe
primary
sequence analysis,wechangedthe sequences ofboth thebulge
and the loop to their comple-mentsin the mutant TM26 (the UCU bulge was changedtoAGA, and the loop sequence CUGGGA was
changed
toGACCCU; Fig.
4). The level ofprotein
binding
to TM26RNAwas
markedly
lower than thatforwild-type
TAR RNA(Fig. 5,
lane 15). Since theloop
is not essential for theon November 10, 2019 by guest
http://jvi.asm.org/
G G
U G U
C A C A
C G C G
G C G C
A U A U
G C G C
U C U U C U
A U A U
G C G C
A U A U
G G C G C G
U G C G C G
C A G C A
G C G C G C
A U A U
U A U A
U A U A
G C G C
G U G U
U A U A
C G C G
U G U G
C G C G
U A U A
C C
U A U A
G C G C
G C G C
TM12 TM27 TM30
-13.1 -13.8 -21.9
G G U G C A C G G C A U G C A U G C A U C G C G A G C A U U A U A G C G U U A C G U G C G U A C U A G C G C TM29 -31.0 U C A U G C A U C G C G A G C A U U A U A G C G U U A C G U G C G U A C U A G C G C TM31 -18.0 A A U C G G U G C A C G G C A U G C U C U
U A U
G C G C
A U A U
C G C G
C G C G
A
G C G C
A U A U
U A U A
U A U
G C G
G U C U C U C U G G TM40 TM38 -6.4 -10.4
U G G G G G G U
C G U G U G U G C G
G G C A C A C A G G
C G C G
U A G C
C A U
C G G C
U A U
C G C
U U
U A A U
C G G C
U A A U
G C C G
G C C G
A A
C G G C
U A A U
A U U A
A U U A
C G C
U G U
A U A U
G C G C
G U G U
G C G C
A U A U
C C
A U U A
C G G C
G C G C
TM 18 TM22 -24.8 -95.0 FIG. 4. Computer-predicted secondary substitutions are boldface.
C G C G
G C G C
A U A U
G C G C
U U A
C C
U U
A U U
G C C G
A U U A
C G G C
C G G C
A A
C G G C
U A A U
A U U A
A U U A
C G G C
U G U
U A U A
C G C G
U G U G
C G C G
U A U A
C C
U A U A
G C G C
G C G C
TM23 TM24 -24.2 -24.9 C G U A C C G U A C G U A U G C A U C G C G A G C A U U A U A G C G U U A C G U G C G U A C U A G C G C TM25 -25.3 A G C C C U U C
C G C G
G C G C
A U A U
G C G C
A U G C A U A U G C A U C G C G A G C A U U A U A G C G U U A C G U G C G U A C U A G C G C TM26 -25.0 A U G C A A U C G A G G C A U U A U A G C G U U A C G U G C G U A C U A G C G C TM37 -17.9
structures and free energies (in kilocalories permole) of TAR RNA and TARmutants. Base
binding of this protein, the reduction inbindingismostlikely due to the changes in the primary sequence of the bulge, particularly the U22-to-A22 alteration. It appears then that the binding site of this nuclear protein overlaps with the binding siteof Tat(23).
TAR RNA point mutations. Having established that the primary sequence ofstem region III is important to TAR
RNAproteinbinding, we wished todetermine whether the secondary structure ofthis region affects protein binding. We therefore constructed a mutant, TM37, containing the singlebase substitutionofC18 toA18, which ispredictedto selectively disruptthe basepairinginstemregionIII andto position a guanidine residue opposite the single adenine residue at position +16 (Fig. 4). Protein binding to this G G U G C A C G G C A U G C U C U A U G C A U C G C G A G C A U U A U A G C G U U A C G U G C G U A C U A G C G C TAR -25.0 G G G U A C G G C G G C A U G C U C U A U G C A U C G C G G C A U U A U A G C G U U A C G U G C G U A C U A G C G C TM39 -28.3 A
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.126.505.76.622.2]<
, ~
~,"e,
r"r"
rwMWNw -.. - .OM- w *g N-W
RNase
dlgestlon
Droducts_
2 3 4 5 6 7 8 9 10 12 13 14 5 6 7
FIG. 5. RNaseprotectionmobility shift assay of TAR RNAmutants.The appearance of theslightlyfaster-migratingband beneaththe majorband(arrow) varied between nuclearextractpreparationsand doesnotexhibitadifferentbinding
specificity.
This minorcomplexis probablyapartialproteolytic digestion product of the majorcomplex. -NE,nonuclearextract.mutant wasundetectable
(Fig. 5,
lane 16). This indicatesthat thesecondarystructureofstemregion
III and thecontextureofthe
unpaired
adenine residue(A16)
play
important
roles intheRNA-proteincomplex formation.Tofocusonthe roleof
the
single bulged
adenine residue atposition
+16,
thisnucleotide was deleted in TM39
(Fig.
4).Binding
activity
was reduced with this molecule (Fig. 5, lane 17). The RNA-protein band seen with TM39,however,
was not asimple quantitative reduction in
signal
intensity. Rather,
it appears that there has been a selective loss ofbinding,
compared with the wild type. This suggests that the TARRNA-binding
protein is a protein complex rather than asingle
polypeptide and that one of theproteins
of thecomplex
requires
contactwith thebulged
adenineresidueatposition +16.
Notably,
theunpaired adenineatthisposition
ishighlyconserved among HIVisolates (15).Competition assay. Tofurther
investigate
theroles ofthe primarysequenceandsecondary structureofstemregionIII inhost cellfactor binding, weperformed competitionassays with wild-type TAR and the mutants TM24and TM37. In theseassays, 0.1, 0.5, and 1.0jig
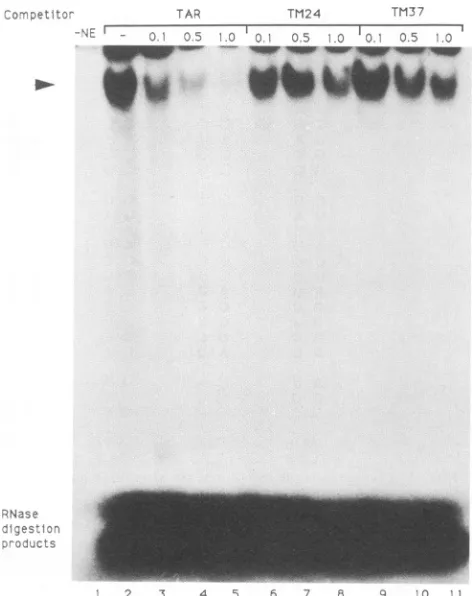
(approximately 100-, 500-, and 1,000-fold molar excesses, respectively) of nonradiola-belled competitor RNA were incubated with the nuclear extract for 10 min prior to the addition ofthe32P-labelled
TAR RNA. Protein binding was efficiently inhibited by competition withthewild-typeTAR RNA(Fig. 6, lanes 3, 4, and 5), whereas the TM24 and TM37 transcripts were relatively inefficient competitors (Fig. 6, lanes 6, 7, and 8
[TM24],
and lanes 9, 10, and11[TM37]).Thesecompetitionassays demonstrate that alterations of either the primary sequence or the secondary structure of TAR RNA stem region III result in a less suitable substrate for host cell proteinbinding.
UVcross-linking. The molecular mass of the TAR
RNA-Co-:et1toQ TAR TM24 TM37
D.1 .5 0 0.0 0.5 .0 o0.1 0.5 1.0 MW 14wME q RNIN
2Nase
," gest01
-z-oduc s
2 3 4 5 6 7 5 9 *0
FIG. 6. Competition assays with nonradiolabelledRNA compet-itors. Nuclear extracts were incubated with 0.1, 0.5, or 1.0 ,ug (approximately 100-, 500-, and 1,000-fold molar excesses, respec-tively) of coldRNAcompetitorpriortotheaddition of 32P-labelled TARRNA,asindicated abovethe lanes. -NE,nonuclearextract.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.146.471.82.338.2] [image:5.612.316.552.386.684.2]:.b
5n-~~~44Y*
[image:6.612.116.254.82.260.2]2 3 4 5 -7
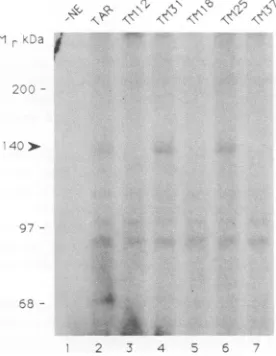
FIG. 7. SDS-PAGE analysis of UV-cross-linked RNA-protein complexes. A 140-kDa protein (arrow) is cross-linked to TAR, TM31, and TM25 RNAs (lanes 2,4,and6)butnot toTM12, TM18,
orTM37 RNA(lanes 3, 5, and 7). -NE, nonuclearextract.
binding protein was estimated by UV cross-linking the 32P-labelled RNAto theprotein and analyzing the products by SDS-polyacrylamide gel electrophoresis (PAGE). A 140-kDa protein (which typically migrated as a doublet) was
cross-linked toTAR RNA and themutantRNAsTM31 and TM25(Fig. 7, lanes 2, 4, and 6), which also bind protein in the gel shift assay, but not to the mutant RNAs TM12, TM18, and TM37 (Fig. 7, lanes 3, 5, and 7), which do not bindprotein in the gel shiftassay.Additionally, the 140-kDa
band was abolished by competition with nonradiolabelled
TAR RNA but notby competition with the mutant RNAs, which bind weakly in the gel shift assay(datanot shown).
DISCUSSION
HIV-1 transcriptional activity is modulated by both viral andhost cell factors. The viralcis-responsive element, TAR RNA, is an essential transcription regulatory element
through which trans-acting factors exert their effect. We haveusedanRNaseprotection mobility shiftassayandUV cross-linking to show thata 140-kDa HeLa nuclearprotein binds specifically to TAR RNA. By mutational analysis of TARRNA,weshow that thesequenceandstructureofstem regionsI, II, and III, the pyrimidine bulge, and the unpaired
adenine (position +16) are important to this interaction. Neither the loop nor stemregion IV, however, is essential for the binding of this host cell protein to TAR RNA. The TAR RNA SBP is distinct inboth its site ofbindingtoTAR RNA and its molecular mass, from previously reported specific TAR RNA-binding cellular proteins (8, 13, 24). Recently, Wu et al. (24) reported two HeLa cell nuclear proteins (TRP-185andTRP-140) which bindto TAR RNA. TRP-185bindingisdependentontheloopsequencesof TAR RNA and requires an additional host cell factor, whereas TRP-140 isapparently nonspecific.
Possible function of TAR RNA SBP. One of the primary effects of Tat is to facilitate the elongation of initiated transcripts whichwould otherwise terminateshortly beyond the 3' end of TAR (7, 10-12, 17, 21). These prematurely terminated transcripts, 55 to 70 bases long, accumulate in uninduced cells and reflect a high rate of nonproductive
initiation events from the HIV promoter(1, 10, 11, 17, 21, 22). Ratnasabapathyetal. (17) have shown thatpositioning
theTAR
region
(orinducer of shorttranscripts)immediately
downstreamofheterologous promoters activates transcrip-tion, causespremature termination, and results in the pro-ductionof shorttranscripts. Since mutations disruptingthe sequence andstructureof the steminhibit theproductionof short transcripts (17, 20, 21), the generation of short
tran-scriptsappearsdependenton theintegrityof the TAR RNA stem. For example, an antisense mutant is not capable of inducingthesynthesisof shorttranscripts (17). Interestingly,
our mutant TM18 (which has an antisense stem) does not support SBP binding. Moreover, the production of short transcripts is independent of the TAR loop (17), as is the
bindingofSBP. SeveralotherTAR stemmutants, reported in the literature, do not support the production of short
transcripts
(11,20).
These correlationssuggest that the SBPis involved in the production of short transcripts from the HIVpromoter.
The sequence and structure of the TAR RNA stem are
remarkably
well conservedamong HIV-1 isolates (2). Thisevolutionary conservation demonstrates that there exists strong selectivepressuretomaintain the
binding
site ofthis cellularprotein. We therefore believe that thisprotein will prove to be animportant
factor in theregulation
ofHIV-1gene
expression.
ACKNOWLEDGMENTS
We thank Judy Mikovits for the HUT-78 nuclear extract and KathyBoris-Lawrie and RamShuklafor valuable discussions.
This work was supported by grant A125531 from the National Institutes of Health and bygrant 000992-7RG from the American Federationfor AIDS Research.
REFERENCES
1. Barry,P.A.,E.Pratt-Lowe,R. E.Unger,and P. A. Luciw.1991. Cellular factorsregulatetransactivationof human immunodefi-ciencyvirus type1.J. Virol. 65:1392-1399.
2. Berkhout, B., A. Gatignol, J. Silver, and K.-T. Jeang. 1990. Efficient trans-activation bythe HIV-2 Tat protein requiresa
duplicatedTAR RNAstructure. Nucleic Acids Res. 18:1839-1846.
3. Berkhout, B., R. H. Silverman, and K.-T. Jeang. 1989. Tat trans-activates the human immunodeficiency virus through a nascentRNAtarget. Cell 59:273-282.
4. Cullen,B. R. 1991. Humanimmunodeficiencyvirusas a proto-typic complexretrovirus. J. Virol.65:1053-1056.
5. Dignam, J. D., R. M. Lebovitz, and R. D. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a
solubleextractfrom isolated mammalian nuclei. Nucleic Acids Res. 11:1475-1489.
6. Dingwall,C.,I.Ernberg,M.J. Gait,S. M.Green,S.Heaphy,J. Karn, A. D. Lowe,M. Singh,M. A. Skinner, and R. Valerio. 1989.Humanimmunodeficiencyvirus1 Tatproteinbinds
trans-activation-responsive region (TAR) RNA in vitro. Proc. Natl. Acad. Sci. USA 86:6925-6929.
7. Feinberg,M.B.,D.Baltimore,and A. D. Frankel.1991.The role ofTat inthehumanimmunodeficiency virus lifecycleindicates
aprimaryeffectontranscriptionalelongation.Proc.Natl. Acad. Sci. USA 88:4045-4049.
8. Gatignol, A.,A.Buckler-White,B.Berkhout,and K.-T.Jeang.
1991. Characterization ofahuman TAR
RNA-binding
protein
thatactivates the HIV-1LTR. Science251:1597-1600. 9. Hart,C.E.,C.-Y.Ou, J.C.Galphin,J.Moore,L. T.Bacheler,J. J.Wasmuth,S. R.Petteway, Jr.,andG. Schochetman. 1989. Humanchromosome12isrequiredforelevated HIV-1 expres-sion in human-hamsterhybridcells. Science 246:488-490. 10. Kao,S.Y.,A.F.Calman,P. A.Luciw,and B. M.Peterlin.1987.
on November 10, 2019 by guest
http://jvi.asm.org/
Anti-termination of transcriptionwithin the long terminal repeat of HIV-1 bytatgeneproduct. Nature(London) 330:489-493. 11. Laspia, M. G., A. P. Rice, and M. B. Mathews.1989. HIV-1Tat
proteinincreases transcriptional initiation and stabilizes elonga-tion. Cell 59:283-292.
12. Marciniak, R. A., B. J. Cainan, A. D. Frankel, and P. A. Sharp. 1990. HIV-1 Tat protein trans-activates transcription in vitro. Cell 63:791-802.
13. Marciniak, R. A., M. A. Garcia-Blanco, and P. A. Sharp. 1990. Identification and characterization of a HeLa nuclear protein that specifically binds to the trans-activation response (TAR) element of human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 87:3624-3628.
14. Muesing,M.A., D.H.Smith, and D. J. Capon.1987.Regulation of mRNA accumulation by a human immunodeficiency virus trans-activator protein. Cell 48:691-701.
15. Myers, G., J. A. Berzofsky, B. Korber, R. F.Smith, and G. N. Paviakis (ed.). 1991. Human retroviruses and AIDS 1991. Los Alamos National Laboratory, Los Alamos, N. Mex.
16. Newstein, M., E. J. Stanbridge, G. Casey,and P. R. Shank. 1990. Human chromosome 12 encodes aspecies-specific factor which increases human immunodeficiency virus type 1 tat-mediated transactivation in rodent cells. J. Virol. 64:4565-4567. 17. Ratnasabapathy, R., M. Sheldon, L. Johal, and N. Hernandez.
1990. The HIV-1 long terminal repeat contains an unusual element thatinduces the synthesis of short RNAs from various mRNAand snRNA promoters. Genes Dev. 4:2061-2074. 18. Rosen, C. A. 1991. Regulation of HIV gene expression by
RNA-protein interactions. Trends Genet. 7:9-14.
19. Roy, S., U. Delling, C.-H. Chen, C. A. Rosen,and N. Sonenberg. 1990.Abulge structure in HIV-1 TAR RNA is required for Tat
binding and Tat mediated trans-activation. Genes Dev. 4:1365-1373.
20. Roy, S.,N. T.Parkin,C.Rosen, J.Itovitch,and N.Sonenberg. 1990. Structural requirements for trans activation of human immunodeficiency virus type 1 long terminal repeat-directed gene expression by tat: importance of base pairing, loop se-quence, and bulges in the tat-responsive sequence. J. Virol. 64:1402-1406.
21. Selby,M.J., E. S. Bain, P. A. Luciw, andB. M. Peterlin.1989. Structure, sequence, and position of the stem-loop in TAR determinetranscriptional elongation by Tat through the HIV-1 longterminal repeat. Genes Dev. 3:547-558.
22. Toohey, M. G., and K. A. Jones. 1989. In vitro formation of short RNA polymerase IItranscripts that terminate within the HIV-1 and HIV-2 promoter-proximal downstream regions. Genes Dev. 3:265-282.
23. Weeks, K.M., C. Ampe, S. C. Schultz, T.A.Steitz,and D. M. Crothers.1990.Fragments of the HIV-1 Tatproteinspecifically bind TAR RNA. Science 249:1281-1285.
24. Wu, F., J. Garcia, D. Sigman, and R. Gaynor. 1991. Tat regulatesbinding of the human immunodeficiency virus trans-activating region RNA loop-binding protein TRP-185. Genes Dev. 5:2128-2140.
25. Zapp, M.L., and M. R. Green.1989. Sequence-specific RNA binding by the HIV-1 Rev protein. Nature (London) 342:714-716.
26. Zucker,M.1987.PCFOLD: version 4.0, RNA secondary struc-tureprediction. National ResearchCouncil of Canada, Ottawa, Canada. [D. Turner, et al. 1987. Cold Spring Harbor Symp. Quant. Biol. 52:123. (Update by J. A.Jaeger.)]