JOURNALOFVIROLOGY, June 1994,p. 3857-3867
0022-538X/94/$04.00+O
Copyright © 1994, AmericanSociety forMicrobiology
A
Nonstructural gag-Encoded
Glycoprotein
Precursor Is
Necessary
for
Efficient
Spreading
and
Pathogenesis
of
Murine
Leukemia
Virusest
ANTOINE CORBIN,' ANNECATHERINE PRATS?2 JEAN-LUC DARLIX,3 ANDMARC
SITBON't*
Laboratoire d'Oncologie CellulaireetMoeculaire, INSERM U363, Institut Cochin de GC&etique Mokculaire, Universite Paris V, 75014Paris, Laboratoire de Biologie Moleculaire du CentreNationalde laRecherche
Scientifique, 31062 Toulouse,2 and LaboRetroINSERM, EcoleNormale Superieure de Lyon, 69364 Lyon Cedex 7,3 France
Received2 December 1993/Accepted8 March 1994
InadditiontotheGag-Pol and Envprecursorswhose translation initiates atAUGcodons, murine, feline, and simiantypeConcoviruses alsoexpressglycosylated Gag-Pol precursors(glycoGag). glycoGag translation is initiated atCUG codons locatedupstream oftheGag AUG initiation codon. In contrast to Gag,glycoGag is translocated into the endoplasmic reticulum and isabsent from virions. Since glycoGag has been described
tobedispensable exvivo, we investigated the in vivo effects ofaglycoGag- mutation in the Friend murine leukemia virus (F-MuLV). F-MuLV induces severeearly hemolytic anemia and subsequent erythroleukemia within 2months afterinoculation of newborn mice. We obtainedaglycoGag- F-MuLV, strain H5, by inserting anoctanucleotide linker downstream of the CUG codonleadingtothereading ofastopcodon in all reading frames upstreamof the Gag AUG. F-MuLV H5 did not inducesevereearly hemolyticanemia, and latency of erythroleukemiawassignificantly increasedmostlikelybecause ofanapproximately 1-week delay in the in vivo spreading. Accordingly, induction of recombinant polytropic viruses was also significantly delayed. Close examination ofexvivo spreading kinetics also showed a slower dissemination of F-MuLV H5. Western blot (immunoblot) performed afterinoculation of newborn mice with this glycoGag- virus indicated theemergence ofnewglycoGag+ viruses. PCR analyses with F-MuLV-specific primers demonstrated in vivo pseudoreversions restoring the glycoGag reading frame. Our results demonstrated that glycoGag expression is positively selected and essential for full spreading and pathogenic abilities.
Lentiviruses, spumaviruses, and oncoviruses from the hu-manT-cell leukemiavirus-bovine leukemia virusgroupexpress nonstructuralproteins which areproducedthrough additional splicings or alternative initiations of translation of the env subgenomic mRNA. Most of these proteins are not incorpo-ratedinto virions,seem tobe dispensable for viral replication, andarethoughttobeinvolved inthe control of viral expression (7). As opposed to these viruses, which are designated as complex, mostof the oncoviruses are designated assimple, in thattheyarebelievedtolack nonstructuralregulatory proteins, although certain groupsamong the latter also encodea setof proteins which are not incorporated into virions. Thus, the Grosscell surfaceantigen(GCSA), initially identified antigeni-cally, is membrane associated and is specifically expressed in cells infected with murine leukemia viruses (MuLV) (26) and is notpresent inthe virions (10, 29). This antigen reactswith antibodies directed against the different gag-encoded viral proteins (MA, p12, CA, and NC) (11, 21, 22, 35, 36, 43, 46), and it has been shown that GCSA and the canonic Pr659ag polyprotein precursor are two alternative translational
prod-ucts of thegag open reading frame which are processed via completelyseparate pathways (10, 13). The canonicPr65-¢gof MuLV anchors tothe internal side of membranes through its N-terminal myristoyl moiety, whereas the GCSA is cotransla-tionally translocated into the endoplasmic reticulum as an
*Corresponding author. On sabbatical leave at the Infectious DiseaseLaboratory,The SalkInstitute, 10010NorthTorreyPinesRd., LaJolla,CA92037-1099. Phone: (619) 453-4100,ext.540. Fax:(619) 554-0341.Electronic mail address: sitbonQasc2.salk.edu.
tDedicated tothememoryofGerard deBilly.
alternative Pr75-"~ precursor.Afterproduction ofan interme-diate gPr80,a8' glycosylated molecule, this alternative P-75~a"
leads to the mature (externallyoriented) plasma membrane-associated gp95-"('' glycoprotein (10, 13, 35). gp951a.- can be cleaved to a gp85,¢at' form lacking NC-related sequences (22, 29). Other membrane-bound amino-terminal fragments (24, 29) and soluble fragments have also been described (10). The GCSA molecular mass is higher than that of the canonic
Pr65,4",4 because of both glycosylations and additional
N-terminal peptidic sequences (11, 35-37). The origin of the GCSA-specific amino-terminal peptide, which has been sug-gested to function as a signal peptide, had been a matter of
controversy. We have previously demonstrated that it is en-coded by the gag open reading frame, from two in-frame alternative CUG initiation codonsupstreamfrom the canoni-calAUG initiation codon ofPr65-4"t4 (31; unpublished data).
Many MuLV which encode GCSA have been described, and cells infected withseveral felineorsimiantype Concoviruses alsoexpressontheirsurfacesgag-relatedglycoproteins (1, 25). Allthese viruses have an open reading frame upstream from
the Gag initiatorwith similargenetic organization and whose deduced amino acid sequences share similar structural
fea-tures. Moreover, it has been recently demonstrated that
hu-manimmunodeficiency virus type 1-infectedcellsalso express
at their surfaces large glycosylated proteins recognized by several monoclonal antibodies directed against gag-encoded proteins of human immunodeficiencyvirustype 1 (18, 30, 38).
Selection of these conservedglycosylatedformsofGag,which
wewill referto asglycoGag,amongvarious retroviral species
suggests that these nonstructural proteins are likely to exert
importantfunctionsduringretroviral selection. SeveralMuLV
3857
Vol. 68,No.6
on November 9, 2019 by guest
http://jvi.asm.org/
mutants lacking glycoGag have been derived. These mutants
are replicationcompetent,and exvivostudies failed toreveal
patentphenotypic differenceswiththeirwild-typecounterparts
(15, 37).
Nonstructural lentiviral proteins, although readily
dispens-able for ex vivo replication, have been shown to harbor
functionspoorly evaluated exvivo and which appear toconfer
selective advantages in vivo (19). Since such dispensable
coding features have never been evaluated in small rodent
retroviral models,wesearched toreaddress the importance of
glycoGag in vivo. For this purpose, we derived a
glycoGag-mutant from strain 57 of the Friend-MuLV (F-MuLV), a
retrovirus whichinduces both lytic andleukemogenic diseases
of the erythroid lineage (27, 39, 42). In contrast to the
previously reported ex vivo studies, we observed that the
mutation had major effects on the in vivo phenotype. The
mutant viruswasattenuated for both the lyticand the
leuke-mogenicdiseases, and its in vivo disseminationwasalso clearly
delayed early after infection. Ex vivo reassessment under
sensitive conditions revealed a significant effect of the
muta-tion onviral dissemination in cell cultures. Furthermore, we
observed
systematic
phenotypic
reversion of the mutation invivo. Conservation of the cysteine-rich hydrophobic region
among all the reverted sequences favored a role for this
sequence inglycoGag functions.
MATERIALS AND METHODS
Plasmid construction. The plasmid pMUSA containingthe
wild-type F-MuLV 57 genome was a
gift
of R. Friedrich.pMUSA contains a permuted form of the
proviral
DNA(at
position 2850 of the
genome)
cloned in the EcoRI site ofvectorpBR327. Numberingwasmadein reference to thefirst
nucleotide of R as number 1.
Toobtain the F-MuLVglycoGag DNA, plasmid pMUSA
waslinearizedbypartial digestion with PstIenzymeand
ligated
with the double-stranded octanucleotide 5'-CGCGTGCA-3',
which has Pstlstickyends.Insertion ofthis linker removes the
PstI site andcreatesanMluI site. Amongseveralrecombinants
plasmids, pMUSAC111 wascharacterized foroneinsertion of
the linker in the correct PstI
site,
atposition
564 of theF-MuLV genome. Insertion of the linkerwascheckedbyDNA
sequencing, using the
dideoxy
method. The Saclfragments
(nucleotide
[nt]
-30 to2560)
ofpMUSA andpMUSAC111
containing
the leadersequencesand thegag genesofwild-type
and mutant F-MuLVs,
respectively,
were introduced into vector pSP65 downstream of SP6 promoter, creating theplasmids pMUSAC 113 and 117.
In vitro transcription and translation. Plasmids pMUSAC
113 and 117were linearized
by
BamHI downstream fromthe3' endof the subcloned
fragment.
Capped RNAsweregener-ated in vitro by SP6 RNA
polymerase
as instructedby
themanufacturer (Promega) in the presence of m7GpppG (0.5
mM) in the transcription reaction mixture. RNA transcripts
were quantified by ethidium bromide stainingonagarosegel,
whichalso allowedtestingof theirintegrity. Invitrotranslation
in rabbit reticulocyte
lysate
(Promega) was performed asindicatedin themanufacturer's instructions,in the presence of
[35S]methionine
(Amersham).
Translationproductswereana-lyzed by electrophoresisin asodiumdodecylsulfate(SDS)-8%
polyacrylamide
gel, and then dried gels were autoradio-graphed.Tissue culture and viral stocks. Unless otherwise stated,
Muis dunni (20) and NIH 3T3 fibroblasts were cultivated in
Dulbecco's modified Eagle'smediumwith glutamine (2 mM),
penicillin (50
IUml-'),
streptomycin (50
mgml-'),
and 10%heat-inactivated fetal calf serum. Cell supernatants were
re-moved from subconfluent transfected NIH 3T3 fibroblasts or infected M. dunni fibroblasts 10 to 18 h after the replacement of the medium.These viralstockswerefiltered (pore size,0.45 mm) and stored in aliquots at -60°C. Viral stocks weretitrated
by using the focal immunofluorescenceassay(FIA)previously
described (41) with monoclonal antibodies discriminating
among the envelope glycoproteins of F-MuLV and Moloney
(M)-MuLV (4, 5, 14).
Construction of the glycoGag- H5 cell line. PMUSAC111
DNA(permuted glycoGag- provirus) wasdigested by EcoRI
andreligatedat a DNAconcentration of 100jLg/mlinorderto
obtain concatemers recreating the normal provirus. NIH 3T3
fibroblastswere cotransfected by20 pg ofpMUSAC111
con-catemers per ml and 4 p.g of vector pSV2neo per ml, by the calcium phosphate method (3). G418 (0.5 p.g/ml) was added after 48h. Two weeks after thetransfection, when clones were visible, cells were trypsinized, diluted to a concentration of 2.5 cells per ml and transferredto96-well microplate dishes (100 pl per well). Cells were grown for another 2 weeks before medium (10
RlI)
was collected from each well containing a clone and assayed for reversetranscriptase activity(16).Viral titrationby endpoint dilution. Filtered viral prepara-tion weretitrated by 10-fold dilutionsfrom 10-2to10-8. Each dilution (0.5 ml) was added in duplicate to 24-well tissue
cultureplatescontaining2 x
104
NIH3T3 cells perwell. Cellswere grownfor aweek,andthen medium(10
[lI)
wascollectedfrom each well andassayed forreverse transcriptase activity.
Origin, infection,andclinical evaluation of mice. All
exper-imentation was conducted on ICFW mice, an inbred line
derived from Carworth Farms White outbred mice (47).
Newborn mice at I to 2 days of age wereinoculated
intraperi-toneally with 0.05 ml of viral stock. For studies of in vivo
dissemination, all litters were inoculated at 1 day of age. In
order to assess the induction of early hemolytic anemia
(EHA), blood samplesweretaken underetheranesthesiafrom
inoculatedmice at3-day intervalsfrom 16 to 24 daysofageby
puncture at the retroorbital sinus with 20-ml heparinized capillary tubes (Drummond Scientific Company, Broomall,
Pa.), andhematocrits, thevolumeoferythrocytes expressedas
the percentage of bloodvolume,were determined. The mice
wereregularlyexamined for splenomegaly by palpationunder
ether anesthesia. Micedisplayinggross organenlargementand
moribund animals were bled for the determination of
hema-tocrits prior to sacrifice and autopsy. The diagnosis of
eryth-roleukemia depended on the presence of splenomegaly and
severe anemia, in the absence of lymphadenotrophy or
thy-motrophy.
Dissemination in vivo. M. dunnifibroblastswereplatedat5 X 104 cells per dish in 60-mm-diameter culture dishes. The
following day, just priortoaddition ofsplenocytes,themedium
wasreplaced with4ml offresh medium.Spleens from infected
mice were removed at different ages anddispersedincomplete
Dulbecco modified Eagle medium. Spleen cell suspensions
were washed once and adjusted to
107
live nucleated cellsml-'. Serial 10-fold dilutions of splenocytes were prepared,
and I ml ofcells at 105,
104,
and103
ml-l (forenumeration ofecotropic infectious centers) or
10',
105, and 104 ml-' (forenumeration ofrecombinantpolytropic infectiouscenters)was
added tothe fibroblast monolayersandincubatedat37°C. The
splenocytes were removed the following day, and the
fibro-blasts were permitted togrow to confluency before
quantita-tion of infectious centers by FIAwith monoclonal antibodies
which discriminate among the envelope glycoproteins of
F-MuLV, M-MuLV, andpolytropicviruses (4, 5, 14).
Western blot. After a 3-day cocultivation with
splenocytes
on November 9, 2019 by guest
http://jvi.asm.org/
IMPORTANCE OF NONSTRUCllURAL GLYCOSYLATED FORMS OF Gag 3859
1Pr75
gag
-Pr65
gag
F-MuLV
57
(wt)
F-MuLV
H5
GlycoGag
CGTCTTGTCT CTGCA CATCGTTCTGTGTTGTCTCTGMGACTGTTMIICTGTAMGTCTGAAAACATGGGCCAG
CGTCrTGTCTGCTGCEA 3SCAIGCATCGTTCTGTGTTGTCTCTGMGACTGTTMrnCTGTAmGTCTGMMCATGGGCCAG
R L
VC C
TRA A
SFCVVSVt
L
|STOP|
MGQ
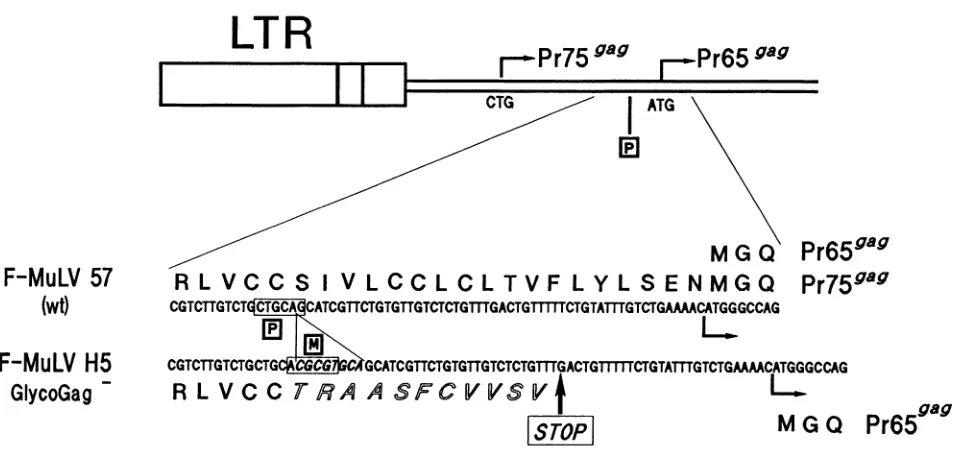
FIG. 1. Schematic representation of the mutated region. (Top) The major CTG initiation codon (nt 355) of the glycoGagprecursor(Pr759ag) and theATGinitiation codon (nt619) of the Gagprecursor(Pr65Mag)areindicated.(Bottom) Nucleotide and amino acidsequencesresultingfrom theframeshifting mutationareindicatedinitalics.P,PstIrestriction site (nt575); M, introduced MluI restriction site.Numbering is performed with the firstnucleotideof Ras 1.wt,wildtype;LTR, long terminalrepeat.
from infected mice (see above), a confluent M. dunni cell
monolayer ina60-mm-diameter dishwaslysedin300 plI of 2x
SDS lysis buffer (135 mM Tris-HCl [pH 6.8], 6% SDS, 20%
glycerol), the lysateswereboiledfor 10min in thepresenceof
5%
13-mercaptoethanol,
and 20pul
wassubjected
toelectro-phoresis in a SDS-10% polyacrylamide gel and immediately
transferred onto a nitrocellulose filter (BA-85, Schleicher &
Schuell) with a high-intensity-field electrotransfer apparatus
(Bio-Rad). Nitrocellulose filterswereblocked byanovernight
incubationat4°CwithTBS-Tween (10mMTris-HCl [pH 7.4],
150mMNaCl, 0.05% Tween 20), 3% nonfat powdered milk,
and 0.02% NaN3. Filters were sequentially incubated with a
rabbitpolyclonalantiserumdirectedagainst theMuLVCAp3O
(1/1,000)andperoxidase-coupled donkeyanti-rabbit
immuno-globulin G antibodies (Amersham Corp.) (1/1000)and
inten-sivelywashed in TBS-Tween before revelation with the
che-moluminescence ECL kit (Amersham Corp.) following the
recommendations of the manufacturer. For Western blots (immunoblots) performed directly on splenocytes, 107 live nucleated cells were resuspended in 100 1.l of phosphate-buffered saline, lysedin 300,ul of 2x lysisbuffer, and treated
asM. dunni cocultivationlysates.
PCR, cloning, and sequence
analysis
of the pseudorevertedregion. Genomic DNAfrom splenocytes of infected mice was
amplified byPCR with the oligonucleotide pairAC6 (nt253 to
273)andC-gag(nt631to651).PCRwasconductedaccordingto
the recommendations of the GeneAmp PCR Reagent kit with
AmpliTaq (Perkin-Elmer Cetus),with final concentrations of 200
mMnucleotides and 2 mM MgCl2, in avolume of 100
[lI.
Thegenomic DNA was subjected to 32 cycles of amplification as
follows: denaturation for 1 minat 96°C, annealingfor 2 minat
39°C,andprimerextension for 2minat72°C.Thesecycleswere
preceded by a 4-min denaturation at 96°C and followed by a
4-min extensionat 72°C. Oligonucleotides (Genset, Paris) used
for PCR were as follows: AC6, 5'-TGATTGA'llIT'ATGCG
CCTGC-3'; and C-gag, 5'-CAAACTI7AAGGGGGTGGTAA
C-3'. The products were digested with the restriction enzymes
PstI orMluI. Aftermigration on a 1.6% agarosegel, the
undi-gested bands were cut out and purified on SpinX columns
(Costar) and reamplified by PCR under thesameconditions. The
PCRfragments, purifiedwith theGene CleanIIkit,wereblunted
bytreatmentwith the Klenowpolymerase, kinased with theT4
polynucleotide kinase, and cloned in a dephosphorylated
M13mp89 phage linearized by EcoRV. After preparation of
single-strandedDNA,10 clones derived from each PCRfragment
were sequenced with the T7 sequencing kit (Pharmacia), by
following the recommendation of the manufacturer and using
either the M13 universal primer, 5'-GTAAAACGACGGC
CAGT-3', orthe AC9 oligonucleotide (nt 421 to 437), 5'-CG
'11lGGACTCl'I'l'GG-3'.
RESULTS
F-MuLVH5, a glycoGag- mutant. We deriveda F-MuLV
mutantwhichdidnotexpressanyglycoGagmolecule.Sinceit
has previously been demonstrated that MuLV-infected cells
express different forms of the glycoGag, which differ intheir N-terminalsequences (35)because of initiationof translation
at alternativeCUG codons(unpublishedobservation),wedid
notchoosetomutatethediverseputative initiation codons of
translation.Rather,weinsertedanoctanucleotideMluI linker
upstream fromthePr65rar region, which resulted ina frame-shift and in the elimination of a PstI site in the specific
glycoGag-coding sequence. This frameshift was predicted to
lead to premature translational arrest in the glycoGag
se-quencewhicheverinitiation codon isused,withoutalteringthe
predicted coding sequence of Pr659ag (Fig. 1). In vitro
tran-scriptionand translation of thegagopenreadingframe ofthe
wild-type plasmid yieldedtwotranslationalproducts,a65-and
a 75-kDa protein characteristic of the wild-type precursors.
Contrastingly, the glycoGag- plasmid gave rise only to the
Pr65rag precursor, with no protein detected in the 70- to 85-kDarange(datanotshown).
Afull-sizeglycoGag- F-MuLVplasmidwascotransfectedin
LTR
lII
iVOL. 68, 1994
I
Pr65g"g
Pr7519
Pr65
989on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.79.556.72.297.2]+ + - - + + Tunica
+ 57 H5 57 H5 57 H5 57 H5
STRAINS INOCULATED
9
16_ t
7!n
4-~
S S a ]glycoGag
66-
_____.4
Pr655
IL-0
LI-wc
--CAp30
29-Lanes 1 2 3 4 5 6 7 8 9 10
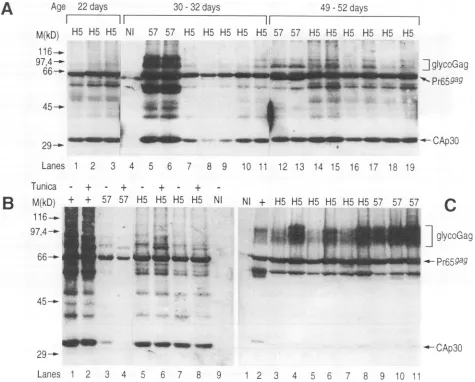
FIG. 2. Gag-related proteins produced byfibroblasts infected with wild-type F-MuLV 57orglycoGag-- F-MuLVH5.M.dunnicellswere
infected with viral stocks derived from the H5 glycoGag- NIH 3T3 clone (lanes 4, 6, 8,and10)or awild-type-infectedNIH3T3 clone(57)
(lanes 3, 5, 7, and 9). Three days later, after tunicamycin (Tunica) treatment(lanes 5, 6,9,and 10)orwithouttreatment(lanes1 to4, 7,
and 8), Gag-related protein expression was determined by Western
blot as described in Materials andMethods, using an CA anti-serum.Lane1,uninfectedM.dunnicells;lane2,controlM. dunni cells chronicallyinfected with F-MuLV 57.MigrationsofglycoGag-specific bands (glycoGag), Gag precursor (Pr65t"i), and CA (CAp3O) are indicated.
F-MuLV 57 F-MuLV H5
(wt
(glycoGag-)
M-MuLV
45 Mos..o.
40
.-
manm.none...
* * u*
*
* a
35-
.-
.
30-
mm.:2 omm
Ems
--
Imean
htc:
32 41 40(± s.e.m.)
(0.8) (0.8) (0.4)FIG. 3. The glycoGag- mutation attenuates the capacity of F-MuLVfor induction of EHA. Newborn ICFW micewereinoculated with thewild-type (wt) F-MuLV57,the glycoGag- F-MuLVH5,or
the M-MuLV. Hematocrits(htc) determinedbetween 19 and 21days ofagefor individual miceareshownasclosedsquares.s.e.m.,standard
errorof themean.
NIH3T3 fibroblasts withaNeor-encoding plasmid,and G418-resistant clonesweretested for viralexpression.The H5clone expressed a high level of virus and was used for all the
subsequent experiments. Gag-related protein expression was tested in this cloneby immunoblotwithananti-CA antiserum. When compared with cells infected with the wild-type
F-MuLV57, several 80- to95-kDa bandswerelackinginthe H5
clone, whereas all the otherproteins were expressed (Fig. 2, compare lanes 4 and 8 with lanes 3 and7).Whentreatingthe
infected cellswith tunicamycin before the assay, we observed that the bands which were present only in the wild-type-infected cells shiftedtoalowerapparentmolecularmass.This
result confirmed that bandspresentonlyin thewild-typevirus
corresponded toglycosylatedforms ofGag.Contrastingly, the
bands presentinboth the wild type and H5 mutant remained
unchangedafter treatment, indicating thatthey corresponded
tounglycosylated
Pr65ag,
CAp3O,andincompletely processed Gag forms, whose expression was not modified inmutant-infected cells (Fig. 2, lanes 6 and 10).
Attenuated pathogenic effects of the glycoGag- mutant of
F-MuLV. Aspreviously describedfor otherglycoGag- MuLV
mutants (15, 37), the H5 clone produced high titers of repli-cation-competent virus (F-MuLV H5) similar to those
ob-tained with the wild-type F-MuLV 57. Mice inoculated as newborns with F-MuLV 57 developed asevere EHA
charac-terized by mean hematocrit below 35%. Contrastingly,
F-MuLV H5 produced onlyverymildEHA, with hematocrits at
leastashighasthoseobserved withthe non-severelyhemolytic M-MuLV(Fig. 3) and F-MuLV B3 (42). F-MuLV also induces
in ICFW mice an erythroleukemia (39, 42) whose latency depends onthe age atinoculation. Although mice inoculated with F-MuLV H5 developed erythroleukemia, latency was markedly delayed when compared with that in F-MuLV 57-inoculated mice, and this delay was more marked in mice inoculated atolderages (Table 1).
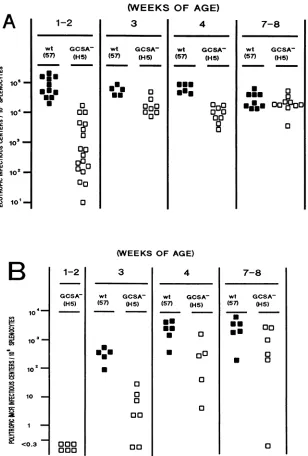
Effects of the glycoGag- mutation on in vivo and ex vivo viral dissemination. Since the glycoGag- mutation
might
attenuate F-MuLVpathogenesis because of an intrinsic flaw
impedingonthe overallspreading of thevirus,we
compared
invivodisseminationofwild-typeF-MuLV57and the
glycoGag-mutant. We observed a highly significant delay in the initial
spreading of the mutant F-MuLV H5
(Fig. 4A).
We alsoobserved that induction of recombinant
polytropic
mink cellfocus-forming
(MCF)
viruseswasalsosignificantly delayed
inF-MuLV H5-inoculated mice compared with that in mice
inoculated with F-MuLV 57 (Fig. 4B). We have recently
demonstrated that vaccination of newborn mice with the
weakly pathogenic F-MuLV B3, which has full
spreading
abilities (40),protects against subsequent
challenges
with thevirulent F-MuLV 57throughinterference to superinfection,a
phenomenon highly dependent onviral dissemination
(8).
Inagreement with a poor initial
spreading
ofF-MuLV H5, weobserved in this system that the glycoGag- mutant failed to
TABLE 1. TheglycoGag mutationdelays onset of F-MuLV-inducederythroleukemia
Leukemialatency (mo[no.ofanimalsl)"
Age(days)of
inoculation" wt57 glycoGag
wt ~~~~~~(H5)
1-2 1.8(19) 3.6(24)
7-10 1.7(6) 3.8(9)
12-13 3.0(16) 5.0(16)
"Micewereinoculated intraperitoneally(1- to 10 day-old mice) orat the retroorbitalsinus(12-to13day-oldmice)with 50p.lofthe F-MuLV viralstocks; wild-type(wt 57) andglycoGag (H5)stockstitratedat1.3x 105 and 1.3x 1t)"
focus-formingunits perml,respectively.
b At leasttwodifferent litters inoculatedatdifferent timeswereused foreach series. All leukemias observedwereerythroleukemias.
M(kD)
>I
45----<
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.316.543.76.304.2] [image:4.612.57.284.78.242.2]IMPORTANCE OF NONSTRUCTURAL GLYCOSYLATED FORMS OF Gag 3861
(WEEKS
OF
AGE)
7-8
wt
GCSA-(57) (H5)
N
(WEEKS OF AGE)
B
8
95 10
10
Z:
E 10
z
g 1
<0.3
1-2
GCSA-(H5)
000 000
3 wt GCSA-(57) (H5)
0 0
DO
4 wt GCSA-(57) (H5)
on
*
* r-2n 0
0
7-8 wt GCSA-(57) (H5)
U.
0
0 *
[image:5.612.163.471.77.533.2]0
FIG. 4. Delayedinvivo dissemination andinduction ofpolytropicrecombinants of theglycoGag mutant F-MuLVH5comparedwith that in wild-type (wt) F-MuLV 57. Mice inoculatedasnewbornswith 50,ulof either thewild-type [wt (57),6.5 x 103focus-formingunitsperanimal)or
the glycoGag- [GCSA- (H5), 6.5 x 104focus-forming unitsperanimal) strainof F-MuLVweresacrificed atdifferent ages. Splenicinfectious
centerswereenumeratedonM. dunni fibroblastsbyFIAwith monoclonal antibodiesspecificforecotropicvirus(H48)orpolytropicviruses(H502 andH514). (A) Ecotropic infectiouscenters;(B) polytropic infectiouscenters.
interfere with F-MuLV 57 hemolytic effects (Fig. 5a) and M-MuLVdissemination (Fig. 5b).
These data on in vivo dissemination, which could at least
partially explain the attenuated phenotype of F-MuLV H5, contrasted withpreviousstudies which failedtopointoutgross defects in the ex vivo dissemination of other
glycoGag-mutants (15, 37). Therefore, we readdressed this question by using our F-MuLV H5 mutant. In agreement with these
previous studies,nodifferenceintheexvivodisseminationwas observedwitharelatively high multiplicityofinfection (MOI)
equal to 1 or 10 (data not shown). On the contrary, we
observed asignificantly delayed initial spreadingof F-MuLV H5 incultureinfectedatalower MOI(lessthan0.01)(Fig. 6).
In vivo phenotypic reversion of the glycoGag- mutation. Because of the discrepancy between the initial spreading impairment of F-MuLV H5 and the normal levels of the late
dissemination,wemonitored cellsinfected with the
glycoGag-H5mutant forpotential reversions. Splenocytesofmice
sacri-ficedatdifferent times afterinoculationwerecocultivatedwith
Dunniindicator cells andanalyzedforGag protein expression.
As revealed with an anti-CA antiserum, we observed a
pro-gressive appearance of bands, which comigrated with the 3
wt
GCSA-(57) (H5)
8308 D0
A
(A
_ 10'
G z
0 03
o
Z102 _
o 104_
Mr. 2
o. 1 10
-4
wt
GCSA-(57) (H5)
on.
O D 0 O 1-2
wt
GCSA-(57) (H5)
.1.
No
8
a 0
°8
(30
DO
0 0
Ocp
00
Obo
0 VOL.68, 1994
on November 9, 2019 by guest
http://jvi.asm.org/
STRAINS INOCULATED
b
M-MuLV infectious centersafter viral inoculations
VACCINATION: NONE
(Day 1) F-MuLVB3 F-MuLV B3 F-MuLV H5 F-MuLVH5 VACCINATION: None F-MuLV B3 F-MuLV H5 CHALLENGE: F-MuLV 57 F-MuLV57
(Day 4)
NONE F-MuLV 57 NONE CHALLENGE: M-MuLV M-MuLV M-MuLV
45
*~~~
a*U:
:--U:
40-
:"mm.
now onmON men U.a
mm BONN me
mmU
m3 . a
ame s~~~~~U
Boom mass~~~~~
30-
25-mean htc: 33.7 (± S.e.m.) (28.5)
38.7 38.3
(28.5) (28.5) 33.3
38.7 (28.5) (28.5)
FIG. 5. The glycoGag- F-MuLVH5 didnotestablishinvivo interference. Micewerevaccinatedat1dayofagewith either of the attenuated F-MuLV H5orthe attenuated F-MuLV B3 and challenged 3 days later with F-MuLV 57orM-MuLV.(A) Lack of protection byF-MuLV H5 against F-MuLV 57-induced EHA. Hematocrits (htc) from mice challenged with F-MuLV 57weredeterminedandareshownasdescribed in the legendtoFig. 4. (B) Lack of efficient interference after F-MuLV H5 vaccination asmeasuredbyinvivo dissemination. Micechallengedwith M-MuLVweresacrificedat21to25daysofage,andthe dissemination ofM-MuLVwasdeterminedbyFIAwithanenvelope-specificmonoclonal antibody (generous giftof B.Chesebro). s.e.m.,standarderrorof themean.
glycoGag-specific bands of F-MuLV 57 controls and which
were absent from Dunni cells infected by the F-MuLV H5 stock used for the inoculation of animals (Fig. 7A). In cocul-tivated Dunni cells treated with tunicamycin, these bands shifted to lower molecular weights, in a pattern identical to
what was observed in F-MuLV 57 controls, confirming that
they corresponded totheGag-related glycoproteins (Fig. 7B).
Wealso verified thisreappearanceofglycoGag directlyonthe
splenocytes of F-MuLV H5-infected mice(Fig.7C).
Since recombinant MCF virusesareinduced after F-MuLV
H5inoculation, althoughwithdelayed kinetics,and since these
viruses maypotentially express glycoGag, itwas possiblethat reappearanceofglycoGag mightbe duetoemergenceofMCF viruses. To address this question,we inoculated F-MuLV H5
intoDBA/2 newborn mice, which donotdevelop MCF virus
infection after F-MuLV inoculation (2, 38a). We observed
emergence ofnew glycoGag-specificbands in the absence of detection of MCF viruses(datanotshown).In agreementwith
thisresult, glycoGag expressiondidnotcorrelate with the level
of MCF virus infection when splenocytes from individual
ICFW mice were assayed for both glycoGag and MCFvirus
presence. We also observed phenotypic reversion of the
gly-coGag- mutant in fibroblast cultures, although MCF virus
induction has never been documented ex vivo. Therefore,
reappearanceofglycoGagwasnotdue solely,ifatall,toMCF
virusemergence.
Sequence analysis ofthe viruses present upon phenotypic
reversion of the glycoGag expression. We amplified the
gly-coGag-specific coding sequences from the genomic DNAof
F-MuLV H5-infected mice splenocytes. Our conditions of
PCR amplificationwere specific for the input virus, sinceno
amplification productwasobserved when splenocytesof
non-infected animals were used. The linker mutation has
elimi-natedthe PstI site and introduceda newMluIsite(Fig. 1).As
expected, PCRamplification products from control F-MuLV
57 mice were readily digested byPstI and resistant to MluI
digestion (Fig. 8,lanes 2 and3). Remarkably,allamplification
E 10
5
10
4
O 10
3lcGg
F-MuLV H5(CA 5 e io wlv-eldseA= 10
w 2 10
DAYS POST-INFECTION
FIG. 6. Dissemination of the wild-type F-MuLV 57 (wt 57) and
glycoGag- F-MuLV HS (GCSA H15) ex vivo. Twelve-well dishes
containing1.5 x 103NIH 3T3 cellswereinfectedby10focus-forming units(1 ml)ofwt57orH5virions. The mediumwascollectedevery
dayfrom 2to9days postinfection,and infectious virusesweretitrated byendpoint dilution, usingareversetranscriptionassayonNIHcells (seeMaterials andMethods).
a
0
0
w
x
1-0
0
0c
0
e
0
o
O..
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.73.543.74.315.2] [image:6.612.317.547.467.655.2]IMPORTANCE OF NONSTRUCTURAL GLYCOSYLATED FORMS OF Gag 3863
Age
22 days
11 1
M(kD)
H5
H5
H5
NI
116 -_
97,4-.s
6-_
_
^of
30
-
32 days
57
57
H5 H5 H5
v _x
45a
29---.
Lanes
1
2
3
4 5 6 7 8 949 - 52
days
I1
H5 H5 57 57 H5 H5 H5 H5 H5
H5
1--t- w v.^
X
]_
^
gglycoGag
_=Pr65ga9g
&:
I
--W4
'-quuFm
-CAp3O
10 11
12 13
14
15 16 17 18 19Tunica
B
M(kD)
116
--97,4-i.
66 -4
- + - + - +- +
-+
+
57 57 H5
H5 H5 H5
NI
Nl
iii
__~~~~~" I...
~~~~~~a
=.-.4V-+
H5 H5
H5 H5 H5H5 57
57 57
H * ,
"s1II.I
29-1
Lanes
1
2
3
4 5 6 7 8 9 1 2 3 4 5 6 7 8 9 10 11FIG. 7. Phenotypic reversionof theglycoGagmutantin vivo. NewbornICFW micewereinoculated with thewild-type (57)orthe glycoGag-(H5) F-MuLV. (Aand B) Mice were sacrificed at differentages (30 days ofage inpanel B), andsplenocytes were plated onM. duilni cell monolayers. After 3 daysofcocultivation, the monolayerswere lysed,andGag-relatedprotein expressionwasdeterminedbyWesternblot with anti-CA antiserumwithout(panelAandpanel B,lanes1, 3, 5, 7,and9)oraftertreatmentwithtunicamycin (Tunica; panel B,lanes2, 4, 6,and 8). (C)Miceweresacrificedat30daysofage,andGag-related protein expressionwasdetermineddirectlyonlysedsplenocytes. NI, splenocytes
orcocultivations derived from noninoculated animals. Control M. dunni cells infectedexvivo with F-MuLV 57wereused(panel B,lanes 1 and
2,andpanelC, lane 2). Otherindicationsare asdescribed in thelegendtoFig.3.
products from F-MuLV H5-infected mice were only partially sensitive to MlulI digestion although were still completely resistant to PstI (Fig. 8, lanes 4 to 11), suggesting that the genomic DNA from F-MuLV H5-infected mice contained bothmutant and pseudorevertant proviruses.
Since we could not exclude partial digestion of the PCR
products, we further analyzed the sequences of two types of amplification products. First, clones were obtained from the
totalamplification materialsdirectlyderived from the infected
mice. Second, clones were also derived from reamplified MluI-resistantDNA in ordertoenrichrevertantDNA(Fig. 9). All of thePCR products obtained from independent amplifi-cations from different mice inoculated with the
glycoGag-mutantwereheterogeneousinsequence.Contrastingly,all the
PCR fragments cloned from the splenocyte DNA of mice inoculated with F-MuLV 57 had the wild-type sequence, indicating that heterogeneity observed in glycoGag- mutant
materialswasnotdue toartefactual misincorporations during
the PCRs. TheglycoGag-coding framewasrestored in all the
cloneswhich differed from F-MuLVH5, indicatingthat rever-tants were selected on glycoGag expression. This restoration
wasalways due to the exact deletionofthe eightnucleotides
which had been introduced inthe wild-type F-MuLV57, and
although the majorityof theclones exhibited additional point mutations, these mutationswere alwayspresent in the
imme-diatevicinity of the deletion.
DISCUSSION
Expressionof nonstructural glycoproteinsencodedin frame
by thegagregionof murine and feline oncovirusesand which are thought tobedispensable for viral replicationwas in fact necessary for complete in vivo spreading and pathogenic
abilities of the F-MuLV. Expression of these proteins was indeed found to be positively selected in vivo. These results wereobtainedby usingaglycoGag-mutantofF-MuLV,strain
A
C
] glycoGag
wr66gag
--CAp30
VOL.68, 1994
--qr-wqw"PrTV
I& 4umwwqomw -ft. qomwNOp im
.,,,
..w", itommiL
Aft.
I
zw"
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.72.547.76.458.2]M(nt)
400
_.,
300
100-O
57
57
H5
H5
H5
H5
-m
mm11
--
r-
III
ND M P M P M P M P M P
[image:8.612.62.295.72.199.2]Lanes
1 2 3 4 5 6 7 8 9 10 11 FIG. 8. Heterogeneity of the mutated region in genomic DNA from miceinoculated with the glycoGag- F-MuLV H5. ICFW mice wereinoculatedasnewbornswith the wild-type F-MuLV 57 (57; lanes 1to3)ortheglycoGag- F-MuLV H5 (H5; lanes4to 11). Genomic DNAwasprepared from splenocytes ofmicesacrificedat32to52days and51 days ofageforstrain H5-and57-inoculated animals, respec-tively. TheglycoGag-specific codingsequenceswereamplified by PCRasdescribed inMaterialsandMethods, andthePCRproductswere digested byMluI(M;lanes2, 4, 6, 8,and 10)orPstI (P;lanes3, 5, 7, 9, and 11). Lane 1, undigested DNA (ND) amplified from mice inoculated withthewild-type F-MuLV 57.Theapproximate length of the DNAfragmentsisindicated.
H5,which lacksexpressionofsuchGag-related glycoproteins
afterintroduction ofan octanucleotide linkerupstream from the ATG initiation codon of the gag sequence. The RNA
region located immediately 5' to the Gag AUG initiator is
highlystructuredandhas been involved inanarrayofcis-and
trans-acting retroviral functions includinggagtranslation,
an-nealing oftRNA, and dimerization and encapsidation of the
genomic RNA into virions (32, 45). Thus, we evaluated the
effectof theglycoGag-mutationontheRNAcisfunctions and
theexpression ofthe othergagproducts. Northern(RNA) blot
analyses performed oninfected cells andonvirionsindicated
thatsimilar levels of the glycoGag- and wild-typevirus RNA
were expressed and encapsidated into virions (data not
shown). This was in agreement with the finding that the
F-MuLV 57-infected clone and the H5 clone yielded similar
levels of virions,asassessed by immunoprecipitationofCAp3O
frompelletedvirions(datanotshown)andby determinationof
infectious titers. Nevertheless, we cannot exclude that
F-MuLV H5-infected cells might produce less Pr65gag than
F-MuLV 57-infected cells (Fig. 2). When monitoring viral
productionin miceinoculatedasnewbornswiththe
glycoGag-mutant,we observedasystematic emergenceof viruses which
had phenotypically reverted to glycoGag expression. These
pseudoreversions might have been selected after mutations
which might have occurred because of transcriptional errors
and/or recombinational events with endogenous sequences.
Remarkably, the reestablishment of the glycoGag-coding
frameinall thepseudorevertantswesequenced,over35 clones
derived from three animals at different ages, was due tothe
exact deletion of the octanucleotide linker. Additional
muta-tions were frequently borne by these sequences but were confined totheclosesurroundings ofthe initial site of
muta-tion. Twocompatible possibilities might explainthat
pseudore-versions were observed only within this limited area. First,
F-MuLV strain Pseudo-reverted sequencesclo-snno.of
(ageofinoc.)
wt 57 R L V
C C
S
I VL C C
LC
(52 days) CGTCT7GTCTGC TGCAGC ATC GTT CTG TGT TGT CTC TGT 9 NA
w
CGTCTTGTCATTTGC AGC ATC GTT CTG TGT TGT CTC TGT 1 1/7
C
CGT CTT GTCTGC TGC A GC ATC GTT CTG TGT TGT CTC TGC 1
FS]
CGT CTTGTCTCGTGCAGC ATC GTTCTGTGTTGT CTC TGT 1
glycoGag-H5 X
(31 days) CGTCTTGTCG.ilTGCAGC ATC GTT CTG TGT TGT CTC TGT 2 _6/11
CGTCTTGTC TCCAGC ATCGTTCTG TGT TGTCZCTGT 1 CGT CTTGTCAfTTGC A GC ATC GTT CTG TGTTGTCTC TGT 1
glycoGag-H5 CGT CTT GTC TGCTGC A GC ATC GTT CTGTGTTGT CTC TGT 1
-(51days)
WIM
[4/5CGTCTTGTC TClGCAGCATC GTTCTGTGT TGT CTC TGT 3
FIG. 9. Pseudorevertedsequences present in mice inoculated with the glycoGag- strain of F-MuLV. Splenocyte DNAs from one52-day-old
mouseinoculated with wild-type(wt)F-MuLV57 andfrom two mice inoculated with glycoGag- F-MuLV H5 sacrificed at either 31or51days
of age were used for analysis ofF-MuLV-specific sequences after PCR amplification and cloning of the PCR fragments. Analyses oftwo independent PCR amplifications performed on the same DNA preparation from the 31-day-old mouse inoculated with glycoGag- F-MuLV H5 are shownseparately.Thewild-typeand the differentpseudoreverted sequences found in each animal are shown with the amino acid and the nucleotide sequencescorresponding to those of the wild type on top. Allpseudorevertedsequences had adeletioncorresponding to the 8-nt linker insertion whichreadilyallowedalignment with the wild-type sequence. The arrow indicates the location of the linker insertion. All othernucleotide
mutations in thepseudorevertedsequences areindicatedin boldface anditalicsand areunderlined,with thecorresponding predictedamino acid. Nonsynonymous amino acid substitutions are indicated with a boxed letter, using the single-letter code. At the end of each line, the frequency at which thecorrespondingsequenceoccurred isindicated, and for each independent experiment the total number ofpseudorevertedsequences(first number)and thetotal number of sequencesanalyzed(second number)areshown in the lastcolumn. NA, notapplicable.
on November 9, 2019 by guest
http://jvi.asm.org/
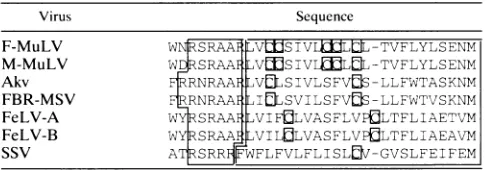
[image:8.612.129.486.396.617.2]IMPORTANCE OF NONSTRUCTURAL GLYCOSYLATED FORMS OF Gag 3865 TABLE 2. Structural conservation of the mutated region in
retroviruses encoding
glycoGaga
Virus Sequence
F-MuLV WNSRAAF.l VWSIVLuLWL-TVFLYLSENM M-MuLV W SRAAF
LVLMSIVLMLL
-TVFLYLSENMAkv
RRNRAAFVLVCLSIVLSFVS
-LLFWTASKNM FBR-MSV FRRNRkAF4ILSVILSFV0
-LLFWTVSKNM FeLV-A w4RSRAAFflLVIFULVASFLVPULTFLIAETVM FeLV-B WY SRAAF LVILLVASFLV0LTFLIAEAVM
SSV ATRSRR
WFLFVLFLISLUV-GVSLFEIFEM
'The sequences arealigned on the methionine initiation codon ofPr65"'5.The arginine-rich and hydrophobic regions are boxed individually; the cysteine residues are boxed. The alignment has been designed to emphasize the con-served distance between thearginine-rich region and the methionine initiator of Pr65¢"`. Other alignments which emphasize the high conservation of twoeight-residue-spaced cysteines within the hydrophobic region could be per-formed.Akv, ecotropic MuLV from AKRmice,FBR-MSV, FBR isolate of the murine sarcoma viruses: FeLV-A and-B,feline leukemia virusessubgroups A and B;SSV,simian sarcoma virus.
similar mutations occurring elsewhere and able to restore a
glycoGagopenreading frame might disruptmoredramatically
either the nucleotidic secondarystructures orthe amino acids
sequences specifically required for this region. Second, this
restricted location for selectedpseudoreversions mightreflect
recombinational events with endogenous sequences which
share a highly conserved glycoGag-encoding region. Indeed,
comparison of the predicted amino acidsequence encodedby
this region in different murine, feline, and simian type C
oncoviruses revealed conserved characteristics. Thus, at the
same location from the AUG initiation codon of the Gag
precursor, these viruses exhibit a cysteine-rich hydrophobic
stretchof20 to23 amino acids whichisimmediately preceded byasmallarginine-rich stretch of6 to7amino acids(Table 2). Interestingly, the amino acid changes observed in the in vivo
pseudorevertants, whichwereall located within the
hydropho-bicstretch, maintainedorevenincreased thehydrophobicityof
thisregion. Inagreementwith afunctionbased on
hydropho-bicity, it has been previously reported that this region mightact
as a signal peptide for translocation into the endoplasmic
reticulum (37). The ratherlong hydrophobic stretch and the upstreamaccumulation of basic amino acids would be unusual
features foracleavable signal peptide oftype I glycoproteins
althoughthese featuresare reminiscent of thesignal peptides
ofmanyretroviral Env precursors which are otherwise type I
glycoproteins (12). However, the overallcharacteristics of the
glycoGag-specific amino-terminal sequence seem tobetter fit
with the description ofthe signal peptide-anchor domain of
type IIglycoproteins(28), i.e., glycoproteinswith acytoplasmic
N-terminal and extracellular C-terminal orientation and an
internal noncleavable signal peptide which acts also as an
anchor domain. Numerous data supportthis hypothesis, such
asthe location of the actualglycosylation sites (11, 35, 36) or
the association of the N-terminal-but not the C-terminal-proteolytically processed fragments of glycoGag with the cell
surface(22, 24, 25, 29). We areinvestigating this question,and
preliminaryresultsindicate thatglycoGagis a type II
glycopro-tein and that the conserved cysteine-rich hydrophobic amino
acid stretch actually acts as a noncleavable signal
peptide-anchor domain
(la).
Although importance ofglycoGagwasunderlinedbythe in
vivomodel,wealso found an effectoftheglycoGag- mutation
on the ex vivo dissemination. The apparent contradiction
between our ex vivo results obtained with the F-MuLV and
those of previous reports on the lack of defect of similar
glycoGag- viruses (15, 37) isnot
likely
tobeduetotheuseofthe M-MuLV in those previous reports, since we have also
observed a lower efficiency of ex vivo spreading with a
gly-coGag-mutantof M-MuLV(not shown).This contradiction is
morelikely relatedtothefact thatdelayinthe
spreading
of theglycoGag-mutant wasapparentonly whenalow
enough
MOIwas used. It is possible that
higher
MOI led to morerapid
emergence of glycoGag+ revertants, similar to what we
ob-served invivo. Delayed dissemination of theglycoGag- virus
was more readily observed in vivo even at a
high MOI,
andinoculated animalsdidnot
develop
severeEHA. SevereEHAinduced after inoculation withthe
fully
virulentwild-type
strainof F-MuLV is no longer observed in 30-day-old animals,
because
hemolysis
is maskedby
more efficient compensatoryerythropoiesis
inolderanimals, notably
because ofastabiliza-tion of their
growth
rates(42). Therefore,
theattenuated EHAobserved in F-MuLV H5-inoculated mice might be due to a
high
level ofviral dissemination inerythroid
percursorsbeing
reached later, when compensatory
erythropoiesis
is not ascrucial for the
growth
of the mice and is more efficient incorrecting
forhemolytic
anemia. The moreslowly spreading
abilities ofthe F-MuLV H5 mutant are also
likely
toexplain
the late appearance of MCF
viruses,
since theirinduction,
which occurs after recombinations between the inoculated
F-MuLV and
endogenous
retroviral sequences, should bedirectly dependent
on F-MuLV dissemination.Accordingly,
we have previously reported that delayed induction of MCF
viruses observed with certain MuLVchimeraswas associated
with adelayed dissemination(33).AsMCF viruses have been
implicated in certain MuLV-induced
leukemogenic
processes(6,
44),
thedelay
inspreading
of F-MuLV H5and/or
MCFviruses might also
explain
the longer latency oferythroleuke-mia induced
by
F-MuLV H5.Nevertheless,
we haverecently
described chimeric viruses between F- and M-MuLV which
also
spread
and induce MCF viruses withdelayed
kineticsclosely
similar to those observed for F-MuLV H5 but whichretain full
erythroleukemogenicity
(33).Also,
the strain B3ofF-MuLV, whichhas full
spreading
abilities andinducesMCFviruses as rapidly as F-MuLV 57, induces
erythroleukemia
aftereven amuchlonger latency
(27,
40,42).
Itis thereforenotexcluded that alteration of
glycoGag expression might
specif-ically influencethe
leukemogenic
effects ofF-MuLV,indepen-dently from its effect on retroviral dissemination. Since
early
events
following
inoculation ofF-MuLVseem toincluderapid
increases of cell
proliferation
in differenthematopoietic
lin-eages
(9, 17, 23),
itmight
be ofinteresttomonitorapossible
role of
glycoGag
in this type ofphenomenon.
In relation tosuch
effects,
we arecurrently
examining
thepossible
effectsofglycoGag
on ex vivo cellproliferation by using
glycoGag-overexpressing
vectors.It seems
paradoxical
that virus-encodedglycoproteins
such asglycoGag, expressed
atthe cellsurface,
are notincorporated
in the virions which are formed
by
budding
at the cellmembranes. Whether
glycoGag
or itsprocessed
derivativesparticipate
in theparticle
formation with concomitantincor-poration
of theenvelope glycoproteins
and theGag
precursor has yet to be evaluated. Nonstructuralexported
proteins
in otherenveloped
viruses have also been described and include the precoreprotein (pre-C)
of thehepadnaviruses,
which share with the retroviruses the use of a reversetranscription
stepduring
thereplication cycle. pre-C
has features which arestrikingly
similartothoseofglycoGag
(34). Thus, pre-C,
which has been described to bedispensable
for viralreplication,
isencoded
by
the coreprotein
openreading
frame from anupstream in-frame initiation codon and is associated with
cellular membranes and is present in secreted forms. In the
VOL.68, 1994
on November 9, 2019 by guest
http://jvi.asm.org/
[image:9.612.66.310.94.179.2]caseof the hepatitis Bvirus, expression of the pre-C proteins,
as HBe antigen, is thought to be related with certain
patho-logical evolution which involves cell proliferation, such as
fibrosis and hepatocarcinoma. The use of animal models will
help in gathering additional information,not readily available
from exvivo studies, onthe role of nonstructural and
appar-ently dispensable regulatory viral glycoproteins.
ACKNOWLEDGMENTS
We are indebted to C. Andre for her help in cloning the PCR products; C. Berlioz for communication of unpublished results; G. de Billy, F. Pozo, and the animal carestaff for excellent assistance; and S.
Gisselbrecht forcontinuous support and helpfuldiscussion.
A.C. is supported by a fellowship from the Fondation pour la RechercheMedicale.
REFERENCES
1. Bergholz,C.M.1981.Synthesis ofvirus-specific proteins in simian sarcomavirus-transformedprimate cells. Virology 114:113-123. la.Berlioz, C., and J. L. Darlix. Unpublished data.
2. Buller, R. S., M. Sitbon, and J. L.Portis. 1988. The endogenous mink cell focus-forming (MCF) gp7O linked to the Rmcfgene restricts MCF virus replication invivo and provides partial resis-tance to erythroleukemia induced by Friend murine leukemia virus. J. Exp. Med. 167:1535-1546.
3. Chen, C., and H.Okayama. 1987.High efficiencytransformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7:2745-2752.
4. Chesebro, B., J.L.Portis,K.Wehrly, andJ. Nishio. 1983. Effect of
murine host genotype on MCF virus expression, latency and leukemia celltype ofleukemias induced by Friend murine leuke-miahelpervirus. Virology 128:221-233.
5. Chesebro, B., K. Wehrly, M. Cloyd, W. Britt, J. Portis,J.Collins, andJ.Nishio. 1981.Characterization ofmousemonoclonal anti-bodiesspecific for Friend murine leukemia virus-induced erythro-leukemia cells: Friend-specific and FMR-specific antigens. Virol-ogy112:131-144.
6. Chesebro, B.,K.Wehrly,J. Nishio, and L. Evans. 1984.Leukemia
inductionbyanewstrain of Friend mink cell focus-inducing virus:
synergistic effect of Friend ecotropic murine leukemia virus. J. Virol. 51:63-70.
7. Coffin, J. M. 1990. Retroviridae and their replication, p. 1437-1500. In B. N. Fields and D. M. Knipe (ed.), Virology, 2nded., Raven PressLtd., NewYork.
8. Corbin, A., and M. Sitbon. 1993. Protection against retroviral diseases after vaccination is conferredbyinterferenceto
superin-fection with attenuated murine leukemia viruses. J. Virol. 67: 5146-5152.
9. Davis, B.R.,B. K.Brightman, K. G.Chandy,and H. Fan.1987.
Characterization of a preleukemic state induced by Moloney murine leukemia virus: evidence fortwoinfection events during leukemogenesis. Proc. Natl. Acad. Sci. USA84:4875-4879.
10. Edwards, S. A., and H. Fan. 1979. gag-related polyproteins of Moloney-murine leukemia virus: evidence for independent synthe-sis ofglycosylated andunglycosylatedforms. J. Virol. 30:551-563. 11. Edwards, S. A., and H. Fan. 1980. Sequence relationship of glycosylated and unglycosylated gag polyproteins of Moloney murine leukemia virus. J. Virol. 35:41-51.
12. Ellerbrok, H., L. d'Auriol, C. Vaquero, and M. Sitbon. 1991. Functional tolerance of the humanimmunodeficiencyvirus type 1
envelope signal peptidetomutations in the amino-terminal and
hydrophobicregions. J. Virol.66:5114-5118.
13. Evans, L. H., S. Dressler, and D. Kabat. 1977. Synthesis and
glycosylation of polyprotein precursors tothe internal core pro-teins of Friend murine leukemia virus. J. Virol. 24:865-874. 14. Evans, L. H., R. P. Morrison, F. G. Malik,J. Portis, and W.J.
Britt. 1990. A neutralizable epitope common to the envelope
glycoproteins of ecotropic, polytropic, xenotropic, and
ampho-tropicmurine leukemiaviruses. J. Virol. 64:6176-6183.
15. Fan, H., H. Chite,E.Chao, and M. Feuerman. 1983. Construction and characterization ofMoloneymurine leukemiavirus mutants
unable to synthesize glycosylated gag polyprotein. Proc. Natl. Acad. Sci. USA80:5965-5969.
16. Goff, S., P. Traktman, and D. Baltimore. 1981. Isolation and properties of Moloney murine leukemia virus mutants: use of a rapid assay for release of virion reverse transcriptase. J. Virol. 38:239-248.
17. Hattori, M., T. Sudo, M. lizuka, S. Kobayashi, S. Nishio, S. Kano, and N. Minato. 1987. Generation of continuous large granular lymphocyte lines by interleukin 2 from the spleen cells of mice infected with Moloney leukemia virus. Involvement ofinterleukin 3. J. Exp.Med. 166:833-849.
18. Ikuta, K., C.Morita, S. Miyake, T. Ito, M. Okabayashi, K. Sano, N. Nakia, K. Hirai, and S. Kato. 1989. Expression of human immunodeficiency virus type 1 (HIV-1) gag antigens on the surface of a cell linepersistentlyinfected with HIV-1 that highly express HIV-1antigen. Virology 170:408-417.
19. Kestler, H. W., III, D.J.Ringler, K. Mori, D. L. Panicali, P. K. Sehgal, M. D. Daniel, and R. C. Desrosiers. 1991. Importance of thenefgenefor maintenance of high virus load and for develop-ment of AIDS.Cell 65:651-662.
20. Lander, M. R., and S. K.Chattopadhyay. 1984. A Mus dunni cell line that lacks sequences closely related to endogenous murine leukemia viruses and can be infected by ecotropic,amphotropic, xenotropic, and mink cell focus-forming viruses. J. Virol. 52:695-698.
21. Ledbetter,J.,and R. C.Nowinski. 1977.Identificationof the Gross cell surface antigen associated with murine leukemia virus-in-fected cells. J. Virol. 23:315-322.
22. Ledbetter,J.,R. C. Nowinski, and S.Emery. 1977. Viralproteins
expressed on the surface of murine leukemia cells. J. Virol. 22:65-73.
23. Mitchell, T. 1993. Increased hematopoiesis in mice soon after infectionby Friend murine leukemia virus. J. Virol.67:3665-3670.
24. Naso, R. B., L. H. Stanker,J. J.Kopchick, V. L. Ng, W. L. Karshin, andR. B.Arlinghaus. 1983. Furtherstudies on the glycosylated
gaggeneproducts of Rauscher murine leukemia virus: identifica-tion of an N-terminal 45,000-dalton cleavage product. J. Virol.
45:1200-1206.
25. Neil, J. C., J. E. Smart, M. J. Hayman, and 0.Jarrett. 1980.
Polypeptides of feline leukemia virus: a glycosylatedgag-related proteinis released into culture fluids.Virology105:250-253.
26. Old, L. J., E. A. Boyse, and E. Stockert. 1965. The G (Gross)
leukemia antigen. Cancer Res. 25:813-819.
27. Oliff,A.I., G. L. Hager, E. H.Chang, E. M.Scolnick,H.W.Chan, andD.R.Lowy. 1980. Transfection ofmolecularlyclonedFriend
murineleukemia virus DNAyields a highlyleukemogenic helper-independent typeC virus. J. Virol. 33:475-486.
28. Parks, G. D., and R. A. Lamb.1991. Topology ofeukaryotictype II membrane proteins: importance of N-terminal positively chargedresidues flanking thehydrophobic domain. Cell 64:777-787.
29. Pillemer, E. A., D. A. Kooistra,0.N. Witte, andI.L.Weissman. 1986. Monoclonalantibody to the amino-terminal L sequence of murine leukemia virus glycosylated gagpolyproteinsdemonstrates their unusual orientation in the cell membrane. J. Virol. 57:413-421.
30. Pinter, A., W.J.Honnen, K. Revesz, and R. Herz. 1992.
Identifi-cation oflarge glycosylated proteins recognized by monoclonal antibodiesagainst HIV-1 gag proteins. AIDS Res. Hum. Retrovi-ruses8:1341-1344.
31. Prats, A. C., G. De Billy, P. Wang, andJ.L. Darlix. 1989.CUG initiation codon used for the synthesis of a cell surface antigen codedby murine leukemia virus. J. Mol. Biol. 205:363-372. 32. Prats, A. C., C.Roy, P. Wang, M. Erard, V. Housset, C. Gabus, C.
Paoletti, and J. L. Darlix. 1990. cis elements and trans-acting
factors involved in dimer formation of murine leukemia virus RNA. J. Virol.64:774-783.
33. Richardson,J.,A.Corbin, F. Pozo, S. Orsoni, andM.Sitbon.1993.
Sequences responsible for thedistinctive hemolytic potentials of Friend and Moloney murine leukemiaviruses are dispersed but confinedtothe T-gag-PRregion. J.Virol. 67:5478-5486. 34. Robinson, W. S. 1990. Hepadnaviridae and their replication, p.
2137-2169.In B. N.Fields and D. M.Knipe, et al.(ed.),Virology,
on November 9, 2019 by guest
http://jvi.asm.org/
IMPORTANCE OF NONSTRUCTURAL GLYCOSYLATED FORMS OF Gag 3867 2nd ed., Raven Press Ltd., New York.
35. Saris, C. J. M., J. van Eenbergen, R. M. J. Liskamp, and H. P. J. Bloemers.1983.Structure ofglycosylated and unglycosylated Gag and Gag-Pol precursor proteins of Moloney murine leukemia virus. J. Virol. 46:841-859.
36. Schultz, A. M., S. M. Lockhart, E. M. Rabin, and S. Oroszlan. 1981. Structure of glycosylated and unglycosylated Gag polypro-teins of Rauscher murine leukemia virus: carbohydrate attach-mentsites. J. Virol. 38:581-592.
37. Schwartzberg, P., J. Colicelli, and S. P. Goff. 1983. Deletion mutants of Moloney murine leukemia virus which lack glycosy-lated Gag protein are replication competent. J. Virol. 46:538-546.
38. Shang, F., H. Huang,K.Revesz,H.Chen,R.Herz, and A. Pinter. 1991. Characterization of monoclonal antibodies against the HIV matrix protein,pl7't"':identification of an epitope exposed on the surface of infected cells. J. Virol. 65:4798-4804.
38a.Sitbon, M., and B. Chesebro. Unpublished data.
39. Sitbon, M., L. d'Auriol, H. Ellerbrok, C. Andre, J. Nishio, S. Perryman, F. Pozo, S. F. Hayes, K. Wehrly, P. Tambourin, F. Galibert, and B. Chesebro. 1991. Substitution of leucine for isoleucine in a sequence highly conserved among retroviral en-velope surface glycoproteins attenuates the lytic effect of the Friend murine leukemia virus. Proc. Natl. Acad. Sci. USA 88: 5932-5936.
40. Sitbon, M.,L.Evans, J.Nishio, K. Wehrly, and B. Chesebro. 1986. Analysis of two strains of Friend murine leukemia viruses differing inability to induce early splenomegaly: lack of relationship with
generation of recombinant mink cell focus-forming viruses. J. Virol. 57:389-393.
41. Sitbon, M., J. Nishio, K.Wehrly, D. Lodmell,and B.Chesebro. 1985. Use ofafocal immunofluorescence assayon live cells for quantitation of retroviruses: distinction of host-range classes in virus mixtures andbiological cloning of dual-tropic murine leuke-mia viruses.Virology 141:110-118.
42. Sitbon, M., B. Sola, L. Evans, J. Nishio, S. F. Hayes, K. Nathanson, C. F. Garon, and B. Chesebro. 1986. Hemolytic anemia and erythroleukemia, two distinct pathogenic effects of Friend-MuLV: mapping of the effects todifferent regionsof the viral genome. Cell 47:851-859.
43. Snyder, H. W.,E.Strockert, andE.Fleissner. 1977. Characteriza-tion of molecularspecies carrying Gross cell surfaceantigen. J. Virol. 23:302-314.
44. Stoye, J. P., C. Moroni,andJ.M.Coffin. 1991.Virologicalevents leading to spontaneous AKR thymomas. J. Virol. 65:1273-1285. 45. Tounekti, N., M. Mougel, C. Roy, R.Marquet, J. L. Darlix, J.
Paoletti, B. Ehresmann, and C. Ehresmann. 1992. Effect of dimerizationontheconformation of theencapsidation Psi domain ofmoloney murine leukemia virus RNA. J. Mol. Biol. 223:205-220.
46. Tung,J.-S., T. Yoshiki, andE.Fleissner. 1976.A corepolyprotein of murine leukemia virusonthe surface ofmouseleukemia cells. Cell 9:573-578.
47. Wendling, F., F. Moreau-Gachelin, and P. Tambourin. 1981. Emergence oftumorigeniccellsduring thecourseof Friend virus leukemias. Proc. Natl. Acad. Sci. USA 78:3614-3618.
VOL.68, 1994