Copyright © 2001, American Society for Microbiology. All Rights Reserved.
The Replicator of the Epstein-Barr Virus Latent Cycle Origin of DNA
Replication,
oriP
, Is Composed of Multiple Functional Elements
MICHELLE D. KOONS, SARAH VAN SCOY,†ANDJANET HEARING*
Department of Molecular Genetics and Microbiology, State University of New York, Stony Brook, New York 11794-5222
Received 9 May 2001/Accepted 9 August 2001
Replication of the Epstein-Barr virus genome initiates at one of several sites in latently infected, dividing cells. One of these replication origins is close to the viral DNA maintenance element, and, together, this replication origin and the maintenance element are referred to asoriP. The replicator oforiPcontains four binding sites for Epstein-Barr virus nuclear antigen 1 (EBNA-1), the sole viral protein required for the replication and maintenance oforiPplasmids. We showed previously that these EBNA-1 sites function in pairs and that mutational inactivation of one pair does not eliminate replicator function. In this study we charac-terized the contribution of each EBNA-1 site within the replicator and flanking sequences through the use of an internally controlled replication assay. We present evidence that shows that all four EBNA-1 sites are required for anoriPplasmid to be replicated in every cell cycle. Results from these experiments also show that the paired EBNA-1 binding sites are not functionally equivalent and that the low affinity of sites 2 and 3 compared to that of sites 1 and 4 is not essential for replicator function. Our results suggest that a host cell protein(s) binds sequences flanking the EBNA-1 sites and that interactions between EBNA-1 and this pro-tein(s) are critical for replicator function. Finally, we present evidence that shows that the minimal replicator oforiPconsists of EBNA-1 sites 3 and 4 and two copies of a 14-bp repeat that is present in inverse orientation flanking these EBNA-1 sites. EBNA-1 sites 1 and 2, together with an element(s) within nucleotides 9138 to 9516, are ancillary elements required for full replicator activity.
The Epstein-Barr virus (EBV) genome is maintained as an autonomously replicating, multicopy plasmid in most latently infected cells (9, 20, 27). Each plasmid is replicated once per cellular S phase, and the progeny plasmids are equally parti-tioned to daughter cells during mitosis (1, 38). Replication and maintenance of the genome during latency require three viral components: an origin of DNA replication, a DNA mainte-nance element (the family of repeats [FR]), and the EBV nuclear antigen 1 (EBNA-1) (reviewed in reference 45). FR is composed of 21 imperfect copies of a 30-bp repeat, 20 of which contain an EBNA-1 binding site (31). In addition to a dimer-ization and DNA binding domain, EBNA-1 contains multiple elements that mediate binding to mitotic host cell chromo-somes (15, 16, 24, 43). It is believed that EBNA-1 performs an essential function in viral genome maintenance through simul-taneously binding the reiterated sites in FR and host cell chro-mosomes, thereby tethering the EBV genome to the host cell chromosomes during cell division (15, 16, 24, 43). The analysis of replicative intermediates by electron microscopy and two-dimensional agarose gel electrophoresis revealed that latent cycle DNA replication can initiate at multiple sites on the EBV chromosome (8, 9, 21, 28). One of these origins is located close to FR, and, together, these two elements are referred to asoriP
(Fig. 1) (8, 44). Recombinant plasmids bearing a 2.2-kb frag-ment of EBV DNA encompassingoriPare replicated in a cell
cycle-regulated manner and maintained efficiently in cells ex-pressing EBNA-1 (47, 48). Thus, smalloriPplasmids provide an experimentally tractable system for studying EBV latent-cycle DNA replication and genome maintenance. Because all of the proteins that are required for the replication of oriP
plasmids, with the exception of EBNA-1, are provided by the host cell, elucidation of the mechanism by which replication initiates atoriPmay provide insight into the nature of mam-malian origins of DNA replication.
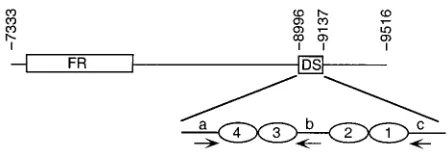
The replicator of oriP is located ca. 1 kb from FR and is referred to as DS due to the presence of a 65-bp dyad symme-try element (10, 28, 32, 34, 46). DS contains four EBNA-1 binding sites arranged as two pairs with 21-bp center-to-center spacing and contains, or is close to, the site at which DNA replication initiates (Fig. 1) (8, 31). Plasmids bearing DS are replicated in an EBNA-1-dependent manner but are not stably maintained in the absence of FR (10, 34, 46). The observation that EBNA-1 differs from eukaryotic viral replication initiator proteins in that it lacks the ability to unwind DNA has led to the hypothesis that EBNA-1 participates in the recruitment of host cell factors required for the initiation of DNA replication from within oriP (7, 11). EBNA-1 interacts with a human single-stranded DNA binding protein (RPA) and a 32-kDa acidic host cell protein (p32/TAP) that may function in latent-cycle DNA replication, but interactions with a DNA helicase or other proteins required for DNA replication have not been reported (41, 42, 49). Data from our previous investigation of the EBNA-1 binding sites within DS showed that the EBNA-1 binding sites function as pairs and that replicator activity was not abolished by the inactivation of one pair of these binding sites (10). This raised the question of why the replicator of the EBV isolate used in our studies (B95-8) contains two pairs of * Corresponding author. Mailing address: Department of Molecular
Genetics and Microbiology, State University of New York, Stony Brook, NY 11794-5222. Phone: (631) 632-8778. Fax: (631) 632-9797. E-mail: [email protected].
† Present address: Department of Pathology, State University of New York, Stony Brook, NY 11794-8691.
10582
on November 9, 2019 by guest
http://jvi.asm.org/
EBNA-1 binding sites. To address this question, we utilized a quantitative short-term replication assay to assess the impact of inactivating individual and paired EBNA-1 binding sites within theoriPreplicator. We found that all four binding sites are required for full replicator activity in an EBV-positive Burkitt’s lymphoma cell line. These results are in agreement with the recently published findings of Yates et al. (46), and our studies further show that the paired binding sites are not functionally equivalent. We also measured the effects of dele-tions withinoriPon replication efficiency, and the results sug-gest that interactions between EBNA-1 and a host cell protein bound to repeats flanking the EBNA-1 binding sites within DS are required for minimal replicator activity. Finally, we rein-vestigated the role of sequences to the right of EBNA-1 bind-ing site 1 and determined that one or more elements within this region are required for fulloriPreplicator function.
MATERIALS AND METHODS
Plasmids.pHEBo-1 and pHEBo-1.1, plasmids bearing EBV (B95-8 strain) nucleotides (nt) 7333 to 9516 and a hygromycin resistance gene (hph) expression cassette, were described previously (11, 37). pHEBo-1.1⌬9138-9516 was derived from pHEBo-1.1 by deleting EBV nt 9138 to 9516 and 56 nonessential nucleo-tides from the 3⬘end of thehphgene by partial digestion withBsaI followed by complete digestion withHpaI. Thehphgene was restored upon ligation of a 471-bp PCR product that contained the appropriatehphsequences followed by
BamHI andHpaI sites. The resulting plasmid contains aBamHI site immediately following EBV nt 9137. pHEBo-1.1⌬9138-9459 was constructed using the same basic strategy; however the PCR product ligated to the digested pHEBo-1.1 fragment contained EBV nt 9459 to 9516. pHEBo-1.1⌬9138-9459⫹M13 was created by introducing a 683-bpBamHI-BglII fragment from M13mp18 at the
BamHI site of pHEBo-1.1⌬9138-9459. pHEBo-2.2 was derived by deleting se-quences (EBV nt 8996 to 9135) between theEcoRV andHpaI sites of pHEBo-1.1⌬9138-9516. pHEBo-2.2.1 was constructed by substituting theEcoRV-HpaI fragment containing DS sequences from pHEBo-1.1(dpm1⫹2) (10) for the small
EcoRV-HpaI fragment of pHEBo-1.1⌬9138-9516. pHEBo-2.2.5, -2.2.6, -2.2.7, and -2.2.8 were created by annealing oligonucleotides with 3⬘complementary ends, synthesizing the complementary strands with the Klenow fragment of
Escherichia coliDNA polymerase I, digesting the double-stranded products with
BamHI and/orBglII, and inserting these fragments at theBamHI site of pHEBo-2.2. pHEBo-pHEBo-2.2.20 and -pHEBo-2.2.24 were generated by PCR amplification of DS sequences using pHEBo-1.1 as the template, digesting the products withBamHI andBglII (pHEBo-2.2.20) orEcoRV andBglII (pHEBo-2.2.24) and inserting the products at theBamHI site of pHEBo-2.2 (pHEBo-2.2.20) or between the
EcoRV andBamHI sites of pHEBo-1.1⌬9138-9516 (pHEBo-2.2.24). pHEBo-1.1 (dpm1), pHEBo-1.1(dpm2), pHEBo-1.1(dpm3), pHEBo-1.1(dpm4), pHEBo-1.1 (dpm1⫹2), pHEBo-1.1(dpm3⫹4), pHEBo-1.1(in1/2[5]), pHEBo-1.1(in2/3[5]), and pHEBo-1.1(in3/4[5]) were described previously (10). pHEBo-1.1(dpm3⫹4; 2⫽HAS) and pHEBo-1.1(dpm1⫹2;3⫽HAS) contain mutations that are pre-dicted to increase the affinity of EBNA-1 for sites 2 and 3, respectively (2). Base pair transitions were introduced at the⫺3,⫹3, and⫹8 positions in site 2 in pHEBo-1.1(dpm3⫹4) or the ⫺4 and ⫹8 positions in site 3 in pHEBo-1.1 (dpm1⫹2) by oligonucleotide-directed mutagenesis as previously described (11). pBS/KS[BclI] was made by inserting aBclI linker into the uniqueXhoI site of
pBluescript KS (Stratagene). ASalI-EcoRV fragment from pHEBo-1.1 contain-ing EBV nt 7333 to 8994 was introduced between the same sites in the polylinker region of pBS/KS[BclI] to create pBS/FR. Similarly, aSalI-BamHI fragment from pHEBo-1.1⌬9138-9516 containing EBV nt 7333 to 9137 was inserted be-tween the same sites in the polylinker of pBS/KS[Bcl] to create pBS/FR.DS. The EBV nucleotides present in each of the plasmids created for this study are given in Table 1. Plasmid DNA was isolated fromdam⫹E. coliDH-1 or DH-5 cells and purified by isopycnic centrifugation on CsCl-ethidium bromide gradients. All plasmids were sequenced after purification to confirm their identity and to confirm that the intended mutations were the only mutations present within DS. Sequence analysis oforiPfrom multiple EBV strains.Whole-cell DNA was prepared from Raji (30), Namalwa (29), FF41 (6), Jijoye (12), and Akata (40) cells as previously described (23). Jijoye and Akata cells were induced to enter the EBV lytic cycle by adding either goat anti-human immunoglobulin M (IgM; Jijoye) or anti-human IgG (Akata) antibodies (HyClone Laboratories, Inc.) to the growth medium at a final concentration of 1%. Whole-cell DNA was isolated after 24 h of incubation in the presence of inducing antibodies. A 782-bp frag-ment of the EBV genome containingoriPDS and flanking sequences (B95-8 nt 8587 to 9363) was amplified by PCR using primers containingEcoRI andHindIII sites. The sequence between nt 8976 and 9142 was determined by direct sequenc-ing of the PCR products or by sequencsequenc-ing pBluescript KS derivatives containsequenc-ing the amplified sequences.
Short-term replication assays.Raji cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (HyClone Laboratories) and 100 U each of penicillin and streptomycin per ml at 37°C. Sixty-one micrograms each of the reference (pHEBo-1 or pKS/oriP) and test plasmid DNAs purified from
dam⫹E. coliwere combined, precipitated with ethanol, and dissolved in 61l of 10 mM Tris-HCl (pH 8.0), 1 mM EDTA (TE). One microliter was reserved for determination of the ratio of test to reference plasmid DNA introduced into cells by quantitative Southern blot analysis. Logarithmic-phase Raji cells were washed with cold growth medium and suspended at 4⫻107cells per ml in cold growth
medium. Electroporations were performed in triplicate by mixing 107cells with
20l of the combined test and reference plasmid DNA. This mixture was transferred to a 0.4-cm-wide cuvette and electroporated with a Bio-Rad Gene Pulser set at 250 V and 960F. Cells were returned to culture in 20% condi-tioned–80% fresh medium and maintained in an actively dividing state for 72 h (three population doublings), at which time low-molecular-weight DNA was isolated from 2⫻107to 5⫻107cells by the method of Hirt (13). Briefly, cells
were washed twice with phosphate-buffered saline, suspended in 2 ml of 10 mM Tris-HCl (pH 8.0)–10 mM EDTA, mixed gently with an equal volume of 10 mM Tris-HCl (pH 8.0)–10 mM EDTA–1.2% sodium dodecyl sulfate and incubated for 20 min at room temperature. High-molecular-weight DNA was precipitated upon the addition of 800l of 5 M NaCl and incubation at 4°C for 12 to 16 h and pelleted by centrifugation at 27,000⫻gfor 30 to 45 min at 4°C. The supernatant was treated with 40g of RNase A per ml for 1 h at 37°C and, subsequently, 10
[image:2.587.51.275.70.148.2]g of proteinase K per ml at 37°C for 1 h. The samples were extracted once with phenol-chloroform-isoamyl alcohol (25:24:1) and once with chloroform-isoamyl alcohol (24:1), and the DNA was precipitated with ethanol. The DNA was pelleted by centrifugation at 25,000⫻gfor 45 min at 0°C, dissolved in 0.3 M sodium acetate (pH 7.5), and reprecipitated with ethanol. The precipitates were FIG. 1. Organization of EBVoriP. The EBV DNA (nt 7333 to
9516) present in pHEBo-1 and pHEBo-1.1 and the locations of FR and DS are shown. Replication initiates within, or close to, DS (8). The arrangement of EBNA-1 binding sites (ovals) and repeat elementsa,b, andc(arrows) within nt 8996 to 9137 are shown at the bottom.
TABLE 1. Description of plasmids constructed for this study Plasmid EBV nucleotides
pHEBo-1.1⌬9138-9516...7333–9137
pHEBo-1.1⌬9138-9459...7333–9137; 9460–9516 pHEBo-1.1⌬9138-9459⫹M13 ...7333–9137; 9460–9516a
pHEBo-2.2...7333–8995 pHEBo-2.2.1...7333–9137b
pHEBo-2.2.5...7333–8995; 9019–9085 pHEBo-2.2.6...7333–8995; 9033–9069 pHEBo-2.2.7...7333–8995; 9019–9069 pHEBo-2.2.8...7333–8995; 9033–9085 pHEBo-2.2.20...7333–8995; 9033–9123 pHEBo-2.2.24...7333–9085
pBS/FR ...7333–8994 pBS/FR.DS...7333–9137
aContains nt 6252 to 6935 from M13mp18 between EBV nt 7333 to 9137 and 9560 to 9516.
bContains base pair transversions at the⫺5 and⫹5 positions of EBNA-1 binding sites 1 and 2.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.587.302.541.555.694.2]dissolved at 2⫻105cell equivalents perl in TE containing 20g of RNase A
per ml.
Restriction enzyme digests containing 2⫻106cell equivalents of
low-molec-ular-weight DNA, 9 U each ofEcoRV andPstI, and 5 U ofDpnI in a solution containing 20 mM Tris-acetate, 10 mM magnesium acetate, 50 mM potassium acetate (pH 7.9), 100 mM NaCl, and 1 mM dithiothreitol (DTT) were incubated for 12 to 16 h at 37°C. Extracts containing pHEBo-2.2-derived plasmids that lack theEcoRV site at EBV nt 8992 were first digested withSacI in 20 mM Tris-acetate–10 mM magnesium acetate–50 mM potassium acetate (pH 7.9)–1 mM DTT and then adjusted to 100 mM NaCl and digested withDpnI,EcoRV, and
PstI as described above. Under these salt conditions, fully methylated plasmid DNA is digested byDpnI while hemimethylated and nonmethylated plasmid DNA is resistant toDpnI digestion (25; M. Dodard and J. Hearing, unpublished data). Digested samples were electrophoresed on 0.8% agarose gels, transferred to Hybond-N⫹(Amersham Pharmacia Biotech), cross-linked to the membrane with UV light, and probed with32P-labeled probe DNA as previously described
(11). A probe comprising EBV nt 8041 to 8996 was used for assays of plasmids derived from pHEBo-1.1 and -2.2, which lack sequences from within DS. A
BamHI-EcoRV fragment from pBS/oriPencompassing EBV nt 7333 to 8992 was used to probe Southern blots of pHEBo-1.1 derivatives bearing point mutations withinoriPDS or deletions to the right of DS. Southern blots for analysis of pBluescript KS-based plasmids were probed with pBS/KS[BclI].
Determination of replication efficiencies.Southern blot quantitation was per-formed with a Molecular Dynamics Storm 860 phosphorimager. The replication efficiencies of test plasmids were calculated as the ratios of replicated test plas-mids to the replicated reference plasmid (pHEBo-1 or pKS/oriP). These ratios were adjusted for the relative amounts of the two plasmids present at the time of electroporation and normalized to the amount of pHEBo-1.1, which was set at 100%. The efficiency with which plasmids replicated per cell generation was calculated using the formulaNt/Nr⫽[(1⫹ε)i⫺(1⫺ε)i]/2i, whereNt/Nris the normalized ratio of replicated test plasmid molecules to replicated reference plasmid molecules aftericell generations andεis the replication efficiency of the test plasmid per cell generation. This calculation assumes that the test plasmid is maintained in dividing cells with the same efficiency as the reference plasmid and that plasmids that are not replicated in one cellular S phase may be replicated in subsequent S phases. It also takes into account the elimination of plasmid DNA
that is never replicated by digestion withDpnI. Statistical analysis of the short-term replication assay data was performed by one-way analysis of variance. Tukey’s “honestly significant difference” test was used to test pairwise compar-isons between the means, andPvalues of⬍0.05 were considered significant.
Long-term plasmid replication and maintenance assays.The efficiency of transformation of Raji cells to hygromycin resistance by pHEBo-1.1 and its derivatives was determined by electroporating 107cells with 2g of test plasmids
as described above. Forty-eight hours after electroporation, cells were plated in 24-well dishes in growth medium supplemented with 300g of hygromycin B (Boehringer Mannheim) per ml at 104, 103, and 102cells per well as previously
described (46). Fresh medium containing 300g of hygromycin B per ml was added to the wells every 6 to 7 days, and the number of wells containing clones was determined 21 days after electroporation. Transformation frequencies were derived by applying the Poisson distribution (36). Two clones which were derived by plating the smallest number of electroporated cells per well were expanded under selection until 4.6⫻106to 1.0⫻107cells were available for analysis.
Low-molecular-weight DNA was isolated as described above and was analyzed as either uncut or digested molecules by Southern blotting. Blots were probed with
32P-labeled pHEBo-1.1 (see Fig. 5) or the large SacII-EcoRV fragment of
pHEBo-1.1 (see Fig. 3).
RESULTS
[image:3.587.134.450.73.346.2]Conservation of sequences inoriPDS.Our previous analysis of EBNA-1 binding sites within oriP DS demonstrated that replicator function was not eliminated by inactivation of two sites provided the remaining sites were separated by 21 bp (10). However, four EBNA-1 binding sites, arranged as pairs with 21-bp center-to-center spacing, are present not only within the B95-8 strainoriPreplicator used in our studies but also within the Raji and C15 EBV genomes (3, 14, 26, 31, 35). To deter-mine if EBV isolates with fewer than fouroriP DS EBNA-1 binding sites exist and to extend the comparisons further into FIG. 2. Sequence oforiPDS and flanking nucleotides from type 1 and type 2 EBV isolates. EBV nucleotides 8977 to 9142 from the B95-8 strain and the same sequence from the Raji, Jijoye, Akata, FF41, and C15 EBV strains are shown. The four EBNA-1 binding sites (boxes) and nonamer repeatsa,b, andc(arrows) are indicated. Differences between the B95-8 sequence and the other viral sequences are underlined. The sequences from the B95-8 and C15 viral genomes were previously reported (3, 35).
on November 9, 2019 by guest
http://jvi.asm.org/
flanking sequences, EBV (B95-8 isolate) nt 8587 to 9363 from four EBV isolates were amplified by PCR and the sequence of nt 8977 to 9142 was determined. These viral sequences were obtained from three Burkitt’s lymphoma cell lines (Raji, Jijoye, and Akata) (12, 30, 40) and a marmoset cell line established by in vitro immortalization of B cells with virus from an infectious mononucleosis patient (FF41) (6). Two EBV types (EBV-1 and -2) have been distinguished (33), and both type 1 (B95-8, Raji, FF41, and Akata) and type 2 (Jijoye) viruses were in-cluded in this comparison.
Two of the viruses examined in this experiment, Jijoye and FF41, are identical to B95-8 between nt 8977 and 9142, while the Raji and Akata viral genomes differ in several places (Fig. 2). These differences were present within two independently derived PCR products and were therefore not the result of polymerase errors during PCR. Raji and Akata viral DNAs differ from each other and the B95-8 sequence at 2 and 3 nt, respectively, flanking the EBNA-1 binding sites. They also differ from B95-8 at 1 nt within EBNA-1 binding site 3, and Akata contains an additional difference within EBNA-1 bind-ing site 2. Neither difference affects the ability of EBNA-1 to bind these sites. In vivo footprinting experiments are consistent with the occupation of site 3 by EBNA-1 in Raji cells (14, 26), and in vitro DNA binding experiments showed that the A-to-G transition within site 2 does not impair EBNA-1 binding (2, 11).
The number and arrangement of DS EBNA-1 binding sites was also found by Snudden et al. to be the same within the genome of a nasopharyngeal carcinoma EBV isolate (C15) (35). The only differences between C15 and the isolates de-scribed here are within EBNA-1 binding sites 2 and 3 (Fig. 2). The A-to-C transversion within site 3, when present in the
context of a high-affinity binding site, has no effect on EBNA-1 binding (2). The A-to-T transversion within site 2 was previ-ously found to reduce the ability of EBNA-1 to bind an oth-erwise high-affinity site (2), but several observations argue that EBNA-1 can bind this site. This nucleotide difference is within only one of the two half sites of the recognition element, and the previous study which showed a reduction in binding affinity contained the transversion at both half sites (2). Also, the binding of EBNA-1 to site 1 facilitates binding to site 2 (11), and this interaction between adjacent EBNA-1 dimers likely stabilizes EBNA-1 bound to site 2 in the C15 genome. In summary, all six of the DS elements examined contain four EBNA-1 binding sites and, additionally, the center-to-center spacing of these sites was conserved among these EBV type 1 and type 2 isolates (Fig. 2).
Contribution of individual EBNA-1 binding sites in DS to replicator activity.The results of this survey suggested that all of the EBNA-1 binding sites within DS, although individually not essential for replicator function, contribute to its activity. Two assays were performed to determine if derivatives of an
oriP-bearing plasmid (pHEBo-1.1) with disabling mutations within individual or paired EBNA-1 binding sites in DS are replicated less efficiently. First, the frequencies with which pHEBo-1.1 and mutant derivatives transformed Raji cells to hygromycin resistance were determined (37, 46). The results of two independent experiments are shown in Table 2. No hygro-mycin-resistant clones were obtained with a pHEBo-1.1 deriv-ative that lacksoriPDS (pHEBo-2.2) at 104cells/well, yielding
a transformation frequency of⬍3⫻10⫺6, while pHEBo-1.1
transformed Raji cells to hygromycin resistance with a fre-quency of 3⫻10⫺2(experiment 1) to 2⫻10⫺3(experiment 2).
[image:4.587.47.543.84.290.2]This high transformation efficiency is due to the ability of TABLE 2. Frequencies of transformation of Raji cells to drug resistance with plasmids bearing wild type or mutatedoriPDS
Expt Plasmid
No. of positive wells/16 wellsgcontaining
indicated no. of cells/well Frequency No. of plasmids/cella 104 103 102
1 pHEBo-2.2 0, 0 N.D.g N.D. ⬍3⫻10⫺6 NAb
pHEBo-1.1 16, 16 16, 16 15, 16 3⫻10⫺2 7.3, 7.0
pHEBo-1.1(dpm1) 16, 16 16, 16 4, 9 5⫻10⫺3 3.3, 3.2
pHEBo-1.1(dpm2) 16, 16 16, 16 6, 12 8⫻10⫺3 5.2, 4.9
pHEBo-1.1(dpm3) 16, 16 15, 16 3, 0 3⫻10⫺3 2.2
pHEBo-1.1(dpm4) 16, 16 16, 16 2, 3 2⫻10⫺3 1.4, 2.2
pHEBo-1.1(dpm1⫹2) 16, 16 16, 16 2, 2 1⫻10⫺3 1.9
pHEBo-1.1(dpm3⫹4) 16, 16 3, 0 0, 1 1⫻10⫺4 2.5, 1.7
pHEBo-1.1(dpm3⫹4;1/2[ATTTA]) 16, 16 3, 0 0, 0 1⫻10⫺4 2.3, 3.1
pHEBo-1.1⌬9138-9516 16, 16 16, 15c 9, 4c 5⫻10⫺3 2.5, 2.5
2 pHEBo-2.2 0, 0 N.D. N.D. ⬍3⫻10⫺6 NA
pHEBo-1.1 16, 16 16, 16 2, 4 2⫻10⫺3d 3.8, 5.9
pHEBo-1.1(in1/2[5]) 16, 16 2, 1 0, 0 1⫻10⫺4 2.3, 6.2e
pHEBo-1.1(in2/3[5]) 16, 16 14, 14 0, 0 2⫻10⫺3d 5.2, 5.3
pHEBo-1.1(in3/4[5]) 16, 16 2, 7 1, 0 3⫻10⫺4 1.8, 2.7f
pHEBo-1.1(dpm3⫹4;2⫽HAS) 16, 16 0, 1 0, 0 3⫻10⫺5 1.5, 1.0
pHEBo-1.1(dpm1⫹2;3⫽HAS) 16, 16 6, 4 0, 0 4⫻10⫺4 6.1,3.0
aAverage number of plasmids per cell for one or two Raji clones derived by transformation with the indicated plasmids. bNA, not applicable; no hygromycin-resistant clones obtained.
cMuch smaller clones 21 days postelectroporation than obtained with pHEBo-1.1.
dpHEBo-1.1 gave rise to more positive wells at both 103and 102cells/well than pHEBo-1.1(in1/2[5]). The calculated frequencies are the same, however, because
the error of this method becomes large when the calculation is based on a small number of negative or positive wells. eDetermination of the copy number for these clones assumed that all the plasmids were dimers of pHEBo-1.1(in1/2[5]). fActual copy number is lower due to the presence of both monomeric and dimeric forms of pHEBo-1.1(in3/4[5]) in these clones. gN.D., none detected.
on November 9, 2019 by guest
http://jvi.asm.org/
pHEBo-1.1 to be maintained as an autonomously replicating plasmid in cells expressing EBNA-1 and is similar to previously reported frequencies (37, 46). Each of the plasmids containing inactivating mutations within one of the four EBNA-1 binding sites transformed Raji cells at reduced frequency compared to pHEBo-1.1, suggesting that all four sites are required for full replicator activity. Because the binding of EBNA-1 to sites 1 and 4 enables binding to sites 2 and 3, respectively (10, 39), we expected pHEBo-1.1 derivatives with disabling mutations within site 1 or site 4 to resemble plasmids with mutations within both sites 1 and 2 or sites 3 and 4 in their ability to trans-form Raji cells. However, pHEBo-1.1(dpm1⫹2) and pHEBo-1.1 (dpm3⫹4) transformed Raji cells at even lower frequencies than pHEBo-1.1(dpm1) and pHEBo-1.1(dpm4) (30- and 300-fold reduction in transformation efficiency, respectively; Table 2).
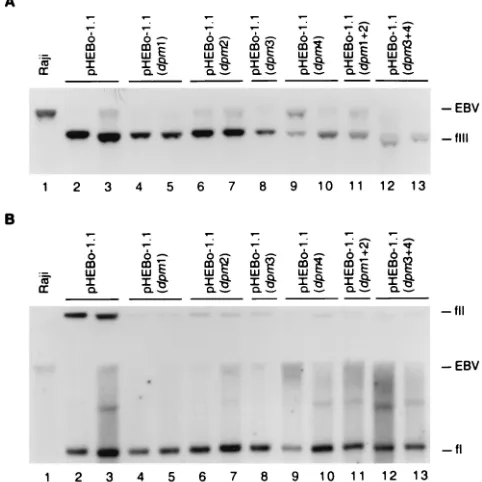
One or two hygromycin-resistant Raji clones established with each plasmid were expanded under drug selection, and the plasmid DNA within the cells was analyzed by Southern blotting. All six plasmids with disabling mutations within indi-vidual or paired EBNA-1 binding sites were present as mono-meric plasmids and at a lower copy number than pHEBo-1.1 (Fig. 3 and Table 2), further supporting the conclusion that all four EBNA-1 binding sites in DS are required for full replicator activity. However, the observation that pHEBo-1.1 (dpm1⫹2) and pHEBo-1.1(dpm3⫹4) were maintained as mo-nomeric circles without gross rearrangements and transformed Raji cells greater than 33-fold more efficiently than an oriP
plasmid which lacks DS demonstrated that plasmids with only one of the two paired EBNA-1 binding sites intact exhibit significant replicator activity, as was previously reported (10). Although wild-type oriP plasmids are replicated once per cell cycle and efficiently maintained in stably transformed cells, these plasmids are rapidly lost during the 2 weeks following introduction into EBNA-1-expressing cells. The establishment of anoriP-plasmid as a stable, autonomously replicating plas-mid is dependent on an undefined epigenetic event that occurs in only a small percentage of the transfected cells (18). Clonal populations of Raji cells harboring derivatives of pHEBo-1.1 with inactivating mutations within individual or paired EBNA-1 sites grew noticeably more slowly than Raji cells containing wild-type pHEBo-1.1 when cultured in the presence of hygro-mycin to select for the presence of the plasmids. The impaired growth of these cells indicated that the mutations affected replicator function and not the epigenetic event that allows an
oriP plasmid to avoid loss from the cell during the several weeks following its introduction. For example, the four cell clones containing pHEBo-1.1(dpm1) and pHEBo-1.1(dpm2) required 32 to 37 days following electroporation to expand for the analysis shown in Fig. 3 and cell clones containing pHEBo-1.1(dpm3) and pHEBo-1.1(dpm4) took 37 to 60 days for this expansion. In contrast, cells harboring wild-type pHEBo-1.1 took only 28 to 30 days of growth before they could be har-vested and analyzed for plasmid copy number.
Mutations that inactivated sites 3 and 4 had a greater effect on transformation efficiency than mutations that inactivated sites 1 and 2, suggesting that the paired EBNA-1 binding sites in DS differ in their contributions tooriP replicator activity. This is not unexpected, as the paired sites differ in sequence context and the relative affinity of the EBNA-1 binding sites. Sites 4 and 3 are immediately flanked by 14-bp inverted
re-peats, each containing the 9-bp repeats referred to by Niller et al. as nonameraandb(26) (Fig. 2). This 9-bp repeat is also found to the right of site 1 (nonamerc) in the same orientation as nonamer b. Differences in the relative affinities of the EBNA-1 binding sites within DS have been detected by an electrophoretic mobility shift assay (2) and by DNase I foot-printing (10, 11, 31). Sites 1 and 4 exhibit the highest affinity for EBNA-1, while site 2 is the lowest-affinity binding site (10, 11). To investigate further the relative contributions of individual and paired EBNA-1 binding sites in DS, the replication effi-ciencies of plasmids bearing mutations in these binding sites were determined using a quantitative, internally controlled replication assay (19) (see Materials and Methods for details). A representative Southern blot used for quantitation of repli-cation efficiency is shown in Fig. 4, and the data are summa-rized in Table 3. In agreement with previous studies, anoriP
[image:5.587.300.541.76.317.2]plasmid lacking DS (pHEBo-2.2) replicated very inefficiently compared to pHEBo-1 (approximately 70-fold lower per cell generation) (10, 32, 34, 46). Mutation of any one EBNA-1 binding site within DS impaired replicator function, and mu-tations in site 3 or 4 reduced replicator activity to a greater extent than mutations in site 1 or 2 (P⬍0.01). Furthermore, inactivation of sites 3 and 4 reduced replicator activity to a FIG. 3. Analysis of plasmid DNA in Raji transformants established with pHEBo-1.1 and its derivatives bearing mutations within single and paired EBNA-1 binding sites. Low-molecular-weight DNA isolated from Raji cells (lane 1) or hygromycin-resistant Raji clones trans-formed by pHEBo-1.1 (lanes 2 and 3), pHEBo-1.1(dpm1) (lanes 4 and 5), pHEBo-1.1(dpm2) (lanes 6 and 7), pHEBo-1.1(dpm3) (lane 8), pHEBo-1.1(dpm4) (lanes 9 and 10), pHEBo-1.1(dpm1⫹2) (lane 11), or pHEBo-1.1(dpm3⫹4) (lanes 12 and 13) was analyzed by Southern blotting with radiolabeled pHEBo-1.1 DNA. The samples were either digested withSacII (A) or analyzed without prior restriction enzyme digestion (B). The positions of covalently closed circular (fI), nicked circular (fII), and linear (fIII) pHEBo-1.1 DNA are indicated on the right. Also indicated are the positions of Raji EBV DNA containing
oriPthat was generated bySacII digestion (A) or mechanical shearing during isolation of low-molecular-weight DNA (B).
on November 9, 2019 by guest
http://jvi.asm.org/
greater extent than inactivation of sites 1 and 2 (P⬍0.01), and this result was consistent with the 10-fold-reduced transforma-tion frequency obtained with pHEBo-1.1(dpm3⫹4) compared to that obtained with pHEBo-1.1(dpm1⫹2) (Table 2). In sum-mary, these results showed that all four EBNA-1 binding sites within DS are required to ensure that replication will initiate once per cell cycle and that the contributions of the two pairs of EBNA-1 binding sites in DS to replicator function are not equivalent.
The sequence context and spatial organization of EBNA-1 binding sites influence their contribution to replicator func-tion.EBNA-1 binds cooperatively to the paired sites within DS in vitro, and double point mutations that eliminate binding to site 4 also eliminate binding to site 3 (10, 39). We therefore expected the replication efficiencies of pHEBo-1.1(dpm4) and pHEBo-1.1(dpm3⫹4) to be the same. However, as suggest-ed by the results of the Raji transformation assay (Table 2), pHEBo-1.1(dpm4) replicated more efficiently than pHEBo-1.1 (dpm3⫹4) (P⬍0.01) and the replication efficiencies of pHEBo-1.1(dpm3) and pHEBo-1.1(dpm4) were the same (P⫽1; Table 3). These results suggest that, in vivo, either EBNA-1 can bind sites 3 and 4 independently or the mutations that eliminate
EBNA-1 binding in vitro do not completely eliminate binding in vivo. Both scenarios could be explained by interactions be-tween EBNA-1 and a host cell protein(s) bound to neighboring sequences in vivo, and interactions between EBNA-1 dimers could also facilitate the occupation of a crippled site. We expected the relative effects of mutations within EBNA-1 bind-ing sites 1 and 2 to be the same as the effects of mutations in sites 3 and 4 but, instead, observed that the mutations within site 2 had a greater impact on replicator function than muta-tions within site 1 (P⬍0.01). One notable difference between sites 1 and 2 is the presence of nonamercadjacent to site 1 and the absence of a similarly positioned nonamer repeat next to site 2 (Fig. 1 and 2). Taken together, these results suggest that interactions between EBNA-1 and a host cell protein(s) bound to the nonamer repeats and between EBNA-1 dimers increase the ability of EBNA-1 to bind sites containing inactivating mutations.
We previously showed that the introduction of 5 or 10 bp between EBNA-1 binding sites 1 and 2 or sites 3 and 4 results in replicators with minimal activity when the other two sites are mutated to prevent EBNA-1 binding (10). These results indi-cated that two DNA-bound dimers of EBNA-1 with a specific spatial arrangement are required for an essential role of EBNA-1 inoriPreplicator function. Because the binding of EBNA-1 to sites in DS does not exhibit the same spatial constraints (10) and because the above results suggested that EBNA-1 interacts with a host cell protein(s) bound to the nonamer repeats, it was of interest to determine if EBNA-1 contributes to replicator ac-tivity when bound to paired sites with altered spacing. pHEBo-1.1 derivatives with 5-bp insertions between adjacent EBNA-1 binding sites {pHEBo-1.1(in1/2[5]), pHEBo-1.1(in2/3[5]), and pHEBo-1.1(in3/4[5])} transformed Raji cells with reduced fre-quencies compared to pHEBo-1.1 (Table 2), and cell clones harboring these plasmids grew more slowly than cells FIG. 4. Short-term replication assay of pHEBo-1.1 derivatives
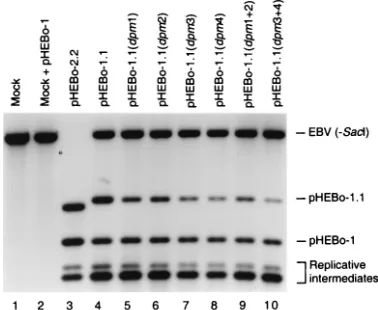
con-taining inactivating mutations within EBNA-1 binding sites. Low-mo-lecular-weight DNA isolated from Raji cells 72 h after electroporation with pHEBo-1 and a test plasmid [pHEBo-2.2 (lane 3), pHEBo-1.1 (lane 4), pHEBo-1.1(dpm1) (lane 5), pHEBo-1.1(dpm2) (lane 6), pHEBo-1.1(dpm3) (lane 7), pHEBo-1.1(dpm4) (lane 8), pHEBo-1.1 (dpm1⫹2) (lane 9), or pHEBo-1.1(dpm3⫹4) (lane 10)] was digested withDpnI,EcoRV, andPstI (lanes 4 to 10) orDpnI,EcoRV,PstI, and
SacI (lane 3) and analyzed by Southern blotting with a probe contain-ingoriPFR. Low-molecular-weight DNA from Raji cells that had not been subjected to electroporation (lane 1) and that had been supple-mented with 400 pg of pHEBo-1 DNA fromdam⫹E. coli(lane 2) was
digested withDpnI,EcoRV, andPstI and included in the analysis. The positions of the diagnosticPstI-EcoRV (2,873 bp) andEcoRV-EcoRV (2,276 bp) fragments derived from pHEBo-1.1 and pHEBo-1, respec-tively, are indicated on the right. Also indicated are two fragments derived from pHEBo-1 and pHEBo-1.1 replicative intermediates con-tainingDpnI-sensitive sites adjacent tooriPFR. EBV (-SacI), 4.4-kb
[image:6.587.68.257.73.228.2]PstI-EcoRV fragment from Raji EBV DNA containingoriPFR. Di-gestion of Raji EBV DNA withSacI, in addition toEcoRV andPstI, results in a 2.7-kb fragment bearingoriPFR which migrates slightly more rapidly than the pHEBo-1.1 diagnostic fragment (lane 3). This digestion allowed for the quantitation ofDpnI-resistant pHEBo-2.2 DNA, which migrates slightly more rapidly than the Raji EBV DNA fragment generated by digestion withEcoRV andPstI (asterisk to the left of lane 3, fragment’s expected position).
TABLE 3. Replication efficiencies of pHEBo-1.1 derivatives with point or insertion mutations inoriPDS
Expt. Test plasmid Repl. eff.a
1 pHEBo-2.2 1.4⫾1.1
pHEBo-1.1 100b
pHEBo-1.1(dpm1) 60⫾2.2
pHEBo-1.1(dpm2) 49⫾3.0
pHEBo-1.1(dpm3) 39⫾3.9
pHEBo-1.1(dpm4) 37⫾3.7
pHEBo-1.1(dpm1⫹2) 51⫾5.2 pHEBo-1.1(dpm3⫹4) 27⫾3.1
2 pHEBo-2.2 2.8⫾2.0
pHEBo-1.1 100c
pHEBo-1.1(dpm3⫹4;1/2[ATTTA]) 26⫾0.81 pHEBo-1.1(in1/2[5]) 42⫾2.7 pHEBo-1.1(in2/3[5]) 57⫾3.2 pHEBo-1.1(in3/4[5]) 44⫾1.9 pHEBo-1.1(dpm3⫹4;2⫽HAS) 29⫾0.43 pHEBo-1.1(dpm1⫹2;3⫽HAS) 54⫾1.7 aReplication efficiencies (repl. eff.) are the percentages of replicated test plasmid DNA relative to replicated reference plasmid (pHEBo-1) DNA, nor-malized to pHEBo-1.1 (100%), per cell generation. The values are means⫾
standard deviations and were derived from three independent determinations. bReplication efficiency over three cell generations relative to pHEBo-1.1 was 141⫾5.1%.
cReplication efficiency over three cell generations relative to pHEBo-1.1 was 102⫾15%.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.587.302.540.487.662.2]taining wild-type pHEBo-1.1. The two cell clones containing pHEBo-1.1(in3/4[5]) grew particularly slowly and required 40 to 45 days following electroporation to expand for the exper-iment shown in Fig. 5 compared to 30 to 31 days for the clones harboring pHEBo-1.1. The slow growth of cells containing these pHEBo-1.1 derivatives under selective conditions for the plasmids is consistent with defects in replicator function. pHEBo-1.1(in1/2[5]) and pHEBo-1.1(in3/4[5]) were reduced in transformation frequency to a greater extent than pHEBo-1.1(in2/3[5]) and also differed in their abilities to be main-tained as monomeric, unrearranged plasmids (Fig. 5). Forms of pHEBo-1.1(in1/2[5]) and pHEBo-1.1(in3/4[5]) with reduced mobility compared to supercoiled pHEBo-1.1 were detected when low-molecular-weight DNA from Raji clones was exam-ined by Southern blotting without prior restriction enzyme digestion (Fig. 5B). Digestion of low-molecular-weight DNA from Raji cells harboring pHEBo-1.1(in1/2[5]) and pHEBo-1.1 (in3/4[5]) with an enzyme that linearizes the founding plasmids yielded plasmid DNA that comigrated with linear pHEBo-1.1 DNA (Fig. 5A), suggesting that the reduced-mobility forms of pHEBo-1.1(in1/2[5]) and pHEBo-1.1(in3/4[5]) are multimers
that were preferentially replicated. Plasmid DNA from one of the two Raji clones established with pHEBo-1.1(in1/2[5]) was recovered inE. coliand, of 18 isolates examined, all were de-termined to be dimers of pHEBo-1.1(in1/2[5]) (data not shown). As predicted on the basis of differences in their transforma-tion frequencies (Table 2), all three plasmids with insertransforma-tions between adjacent EBNA-1 binding sites were reduced in rep-lication efficiency compared to pHEBo-1.1 and pHEBo-1.1 (in2/3[5]) replicated with greater efficiency than pHEBo-1.1 (in1/2[5]) and pHEBo-1.1(in3/4[5]) (P⬍0.01; Table 3). The impact of a 5-bp insertion between EBNA-1 sites 3 and 4 on replication efficiency was much less than the effect of mutating both sites, indicating that EBNA-1 bound to sites 3 and 4 with altered spacing can still contribute to replicator activity (P⬍
0.01). These results lead us to conclude that the spatial ar-rangement of EBNA-1 sites 3 and 4 in Raji cells is important, but not essential. The analysis of the effects of altering the spacing between EBNA-1 binding sites also underscored the functional difference(s) between the paired sites. The replica-tion efficiencies of pHEBo-1.1(in3/4[5]), pHEBo-1.1(dpm3), and pHEBo-1.1(dpm4) did not differ (Pⱖ0.23), while the ef-fect of altering the spacing of binding sites 1 and 2 was greater than the effect of mutating site 1 (P⬍0.01) but not site 2 (P⫽
0.19).
An alternative explanation for these results is that the inser-tion mutainser-tions alter the sequences between the EBNA-1 sites so they can no longer participate in some unknown func-tion such as interacfunc-tion with a host cell protein. To investigate this possibility, the 5 bp between binding sites 1 and 2 were changed from 5⬘-ACCCG-3⬘ to 5⬘-ATTTA-3⬘ in pHEBo-1.1 (dpm3⫹4). The transformation frequency and replication effi-ciency of this plasmid, pHEBo-1.1(dpm3⫹4;1/2[ATTTA]), did not differ from those of pHEBo-1.1(dpm3⫹4) (P ⫽ 1; Tables 2 and 3). Therefore, the replication defects of pHEBo-1.1(in1/2[5]) and, most likely, pHEBo-1.1(in3/4[5]) are due to an alteration in the spacing between the paired EBNA-1 bind-ing sites. The replication defect of pHEBo-1.1(in2/3[5]) is also consistent with a strict spatial requirement for the two paired EBNA-1 binding sites. However, the 5-bp insertion within this plasmid disrupts the 14-bp sequence that is present in inverse orientation flanking EBNA-1 binding site 4. Additional exper-iments will be necessary to determine if the negative impact of the 5-bp insertion between EBNA-1 sites 2 and 3 is due to dis-ruption of the spacing between these sites or the 14-bp repeat.
The low affinity of EBNA-1 binding sites 2 and 3 relative to that of sites 1 and 4 is not required for the paired sites to function in the initiation oforiPplasmid DNA replication.We considered the possibility that the arrangement of higher-affinity (sites 1 and 4) and lower-higher-affinity (sites 2 and 3) EBNA-1 binding sites within oriP DS is important for replicator function. To test this hypothesis, we changed site 2 within pHEBo-1.1(dpm3⫹4) and site 3 within pHEBo-1.1(dpm1⫹2) to high-affinity EBNA-1 binding sites. Base pair transitions were introduced at positions⫺4 and⫹8 within site 3 and at posi-tions⫺3,⫹3, and⫹8 within site 2 (2). No differences in the ability of EBNA-1 to bind the altered sites or the adjacent intact sites in each pair were detected by DNase I footprinting performed with full-length EBNA-1 (data not shown). Both pHEBo-1.1(dpm3⫹4;2⫽HAS) and pHEBo-1.1(dpm1⫹2;3⫽
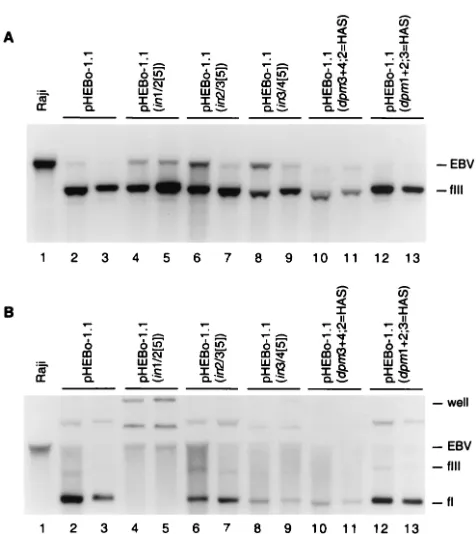
[image:7.587.44.282.67.334.2]HAS) were maintained as monomeric circles (Fig. 5) and FIG. 5. Analysis of plasmid DNA in Raji transformants established
with pHEBo-1.1 and its derivatives bearing insertion and point muta-tions inoriPDS. Low-molecular-weight DNA isolated from Raji cells (lane 1) or hygromycin-resistant Raji clones transformed by pHEBo-1.1 (lanes 2 and 3), pHEBo-pHEBo-1.1(in1/2[5]) (lanes 4 and 5), pHEBo-1.1 (in2/3[5]) (lanes 6 and 7), pHEBo-1.1(in3/4[5]) (lanes 8 and 9), pHEBo-1.1(dpm3⫹4;2⫽HAS) (lanes 10 and 11), or pHEBo-1.1(dpm1⫹2;3⫽ HAS) (lanes 12 and 13) were analyzed by Southern blotting with radiolabeled pHEBo-1.1 DNA. The samples were either digested with
SacII (A) or analyzed without prior restriction enzyme digestion (B). The positions of covalently closed circular (fI) and linear (fIII) pHEBo-1.1 DNA and Raji EBV DNA containingoriPthat was generated by
SacII digestion or mechanical shearing during isolation are indicated on the right. Nicked circular plasmid DNA is present in panel B, lanes 2, 3, 6, 7, 12, and 13, between the wells and the largest viral DNA fragments complementary to the probe.
on November 9, 2019 by guest
http://jvi.asm.org/
yielded replication efficiencies in the short-term replication assay (Table 3) similar to those for pHEBo-1.1(dpm3⫹4) and pHEBo-1.1(dpm1⫹2) (P⫽1). These results indicate that the pairing of higher- and lower-affinity EBNA-1 binding sites is not required for replicator function.
An element(s) located to the right of nonamerccontributes to oriP replicator activity. Previous investigations of the boundaries oforiPrelied on the ability of mutatedoriP plas-mids to replicate and be maintained over many cell generations (4, 32, 44). The reduced copy numbers observed with plasmids lacking the sequence to the right of EBV nt 9134 in several of these studies suggested that sequence elements in addition to EBNA-1 binding sites, though not essential fororiPfunction, may contribute to replicator activity (see Fig. 1 and 2 for the location of nonamer c and nucleotide coordinates) (32, 44). TheoriPdeletion derivatives examined in these previous stud-ies lacked part or all of nonamercin addition to missing all other right-hand EBV sequences. To determine if the se-quence to the right of the EBNA-1 binding sites within DS is required for full replicator activity and to distinguish between a requirement for nonamer c and nucleotides to its right, a pHEBo-1.1 derivative lacking all EBV nucleotides to the right of nonamer c was tested for its ability to be replicated and maintained in Raji cells. This plasmid, pHEBo-1.1⌬9138-9516, transformed Raji cells to drug resistance with sixfold-reduced frequency compared to pHEBo-1.1, and its copy number in two Raji cell clones was also reduced in comparison to that of 1.1 (average of 2.5 copies per cell for pHEBo-1.1⌬9138-9516 compared to 7.1 copies per cell for pHEBo-1.1; Table 2). In agreement with these results, the replication effi-ciency of pHEBo-1.1⌬9138-9516 was only 70% per cell gener-ation (Table 4). The replicgener-ation defect of pHEBo-1.1⌬ 9138-9516 may be due to the loss of an auxiliary element(s) that facilitates the initiation of DNA replication or, alternatively, a negative effect of placing plasmid sequences adjacent to DS (see, e.g., reference 4). To address the latter possibility, the right-hand flanking sequence was changed in two ways. A 683-bp fragment from phagemid M13mp18 was substituted for EBV nt 9138 to 9459 in pHEBo-1.1 and the ability of this plasmid and its parental plasmid lacking M13 DNA (pHEBo-1.1⌬9138-9459) to be replicated was determined. The EBV se-quences present in pHEBo-1.1⌬9138-9516 (nt 7333 to 9137)
were also introduced into pBluescript KS, and the replication efficiency of this plasmid (pBS/FR.DS) was compared to that of pBluescript derivatives which contain EBV nt 7333 to 9516 (pBS/oriP) or EBV nt 7333 to 8994 (pBS/FR). pBS/FR rep-licated very inefficiently, as expected for a plasmid lacking DS. pHEBo-1.1⌬9138-9516, pHEBo-1.1⌬9138-9459, pHEBo-1.1⌬9138-9459⫹M13, and pBS/FR.DS replicated with the same reduced efficiency (approximately 70 to 80% per cell generation), indicating that the reduced replication efficiency of pHEBo-1.1⌬9138-9516 was not likely due to changes in flanking sequences (Table 4). The effect of deleting EBV nt 9138 to 9516 from a plasmid with inactivating mutations within EBNA-1 binding sites 1 and 2 was also examined. If the right-hand sequence contributes to replicator activity independently of EBNA-1 binding sites 1 and 2, the replication efficiency of this plasmid (pHEBo-2.2.1; Fig. 6) would be expected to be lower than the replication efficiency of pHEBo-1.1(dpm1⫹2). The replication efficiencies of these two plasmids, however, were the same (P⫽0.98). Taken together, these results sug-gest that the deletion of EBV nt 9138 to 9459 either disrupts or eliminates an element(s) required for full activity of theoriP
replicator and this putative element functions in conjunction with EBNA-1 binding sites 1 and 2.
Minimal sequence requirements for theoriPreplicator.The results of our mutational analysis of EBNA-1 binding sites (10) showed that the paired sites are essential components of the
oriPreplicator, and data presented here suggest that at least one additional sequence element, located between EBV nt 9138 and 9516, is required to ensure that replication oforiP
plasmids will initiate once per cell cycle. To identify the min-imal fragment fromoriP DS that provides replicator activity and to determine if there are additional, nonessential
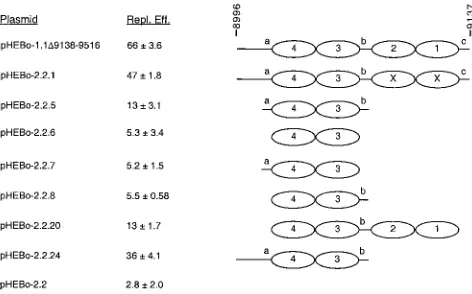
[image:8.587.45.283.91.175.2]compo-FIG. 6. Replication efficiencies of pHEBo-1.1 and -2.2 derivatives with sequences inoriPDS deleted. The replication efficiencies (Repl. Eff.) of pHEBo-1.1⌬9138-9516 and pHEBo-2.2 derivatives lacking se-quences inoriPDS were determined in triplicate and are given as percent per cell generation with standard deviations relative to that of pHEBo-1.1. These plasmids all containoriPFR and the sequences normally present between FR and DS (EBV nt 7333 to 8995; see Fig. 1). The DS-derived sequences present in each plasmid are represent-ed schematically at the right, and the nucleotide coordinates are at the top. Ovals, EBNA-1 binding sites; a, b, and c, nonamer repeats; EBNA-1 binding sites labeled X, sites containing inactivating mu-tations at the⫹5 and⫺5 positions (10). The replication efficiency of pHEBo-1.1 was 102⫾15%, and that of pHEBo-1.1⌬9138-9516 was determined in the experiment presented in Table 4.
TABLE 4. Replication efficiencies of pHEBo-1.1 and pBS/oriP
derivatives with deletions of sequences flanking DS Test plasmid Repl. eff.a pHEBo-2.2 ...0.54⫾0.21 (3) pHEBo-1.1 ... 100 (3) pHEBo-1.1⌬9138-9516 ... 66⫾3.6 (3) pHEBo-1.1⌬9138-9459 ... 72⫾5.1 (3) pHEBo-1.1⌬9138-9459⫹M13 ... 61⫾3.1 (3) pBS/FR... 11, 3.0 (7.9)b
pBS/FR.DS ... 74⫾14 (5) aReplication efficiencies (repl. eff.) are the percentages of replicated test plasmid DNA relative to replicated reference plasmid (pHEBo-1 or pBS/oriP) DNA per cell generation. The replication efficiencies of pHEBo-1.1-derived plasmids are expressed relative to that of pHEBo-1.1, which was set at 100% (122⫾5.1). Unless otherwise indicated, the values are the means⫾sample standard deviations and the numbers of experiments are included in parentheses. bReplication efficiency of test plasmid per cell generation. The values are the results of two independent experiments, and the number in parentheses is the average of the two experiments.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:8.587.303.539.452.599.2]nents that contribute to replicator function, we constructed derivatives of pHEBo-1.1⌬9138-9516 with mutations between EBV nt 8995 and 9137 and analyzed their ability to be repli-cated in the short-term assay. The approach taken was to con-sider the 14-bp repeats flanking EBNA-1 binding sites 4 and 3 (containing nonamersaandb) and nonamercpotential func-tional elements and to test the EBNA-1 binding sites in various combinations with the nonamer repeats and other flanking sequences. We chose to focus on sites 3 and 4 because they contribute more to replicator activity than sites 1 and 2 (Tables 2 and 3). TheoriPDS sequences present in these plasmids are summarized in Table 1 and are shown schematically in Fig. 6. A plasmid containing EBNA-1 binding sites 3 and 4 but lacking all other sequences from DS and right-hand flanking se-quences (pHEBo-2.2.6) resembled the parental plasmid lack-ing DS (pHEBo-2.2) in its inability to be replicated in Raji cells (P⫽1), suggesting that one or more elements, in addition to these EBNA-1 binding sites, are required to constitute mini-mal replicator activity. Inclusion of either of the 14-bp repeats flanking sites 3 and 4 did not give rise to replicator activity (compare pHEBo-2.2 with pHEBo-2.2.7 and -2.2.8;P⫽1), but a plasmid containing EBNA-1 binding sites 3 and 4 and both 14-bp repeats replicated, albeit inefficiently (compare pHEBo-2.2 with pHEBo-pHEBo-2.2.5;P ⬍ 0.01). Inefficient replication was also observed when all four EBNA-1 binding sites and the 17-bp between the paired sites containing nonamer b were present (compare pHEBo-2.2 with pHEBo-2.2.20;P⬍0.01). Replication efficiency increased significantly when the 24 bp between nt 8994 and 9019 were restored (compare pHEBo-2.2.5 with pHEBo-2.2.24;P⬍0.01). The latter result indicates that the inclusion of this sequence either provides an element that enhances the activity of EBNA-1 binding sites 3 and 4 and flanking repeats or places this minimal replicator at an appro-priate distance from an auxiliary element located betweenoriP
FR and DS.
DISCUSSION
Assessment of oriP replicator function. Previous genetic analyses of the EBV oriP replicator carried out by this and other laboratories relied predominantly on quantitation of the ability of mutatedoriPplasmids to transform EBNA-1-express-ing cells to drug resistance and the copy number of the plas-mids in stably transformed cells cultured over many cell gen-erations (4, 10, 11, 22, 32, 44, 46). The transformation assays used in these previous experiments suffered from the absence of an internal control for transfection efficiency. Furthermore, the copy number of anoriPplasmid is determined not only by the replication efficiency of the plasmid but also by the amount of plasmid DNA introduced into a cell at the time of transfec-tion (47). For these reasons, these experimental approaches have provided only a semiquantitative measure of replicator function and have been unable to allow accurate comparisons between plasmids with mutations that reduce but do not elim-inate replicator activity (see, e.g., references 10 and 44). These limitations are highlighted by the results presented in this study. Although the transformation frequencies of plasmids reported here generally reflected relative replication efficien-cies, there were several notable exceptions that suggested the influence of experimental variation [e.g., pHEBo-1.1(dpm3⫹4;
2⫽HAS), pHEBo-1.1(dpm1⫹2), and pHEBo-1.1(in1/2 [5]); Tables 2 and 3]. Note, too, that the plasmid copy numbers for Raji clones transformed by pHEBo-1.1(dpm4) and pHEBo-1.1 (dpm3⫹4) were similar and did not reflect the 20-fold differ-ence in transformation efficiency (Table 2) and different rep-lication efficiencies (Table 3) exhibited by these plasmids. In several previous studies, short-term replication assays were also used to characterize mutatedoriPplasmids. These assays either lacked an internal control for transfection efficiency and experimental error in the isolation of plasmid DNA from transfected-cell populations (10) or compared the amount of replicated plasmid DNA to the total amount of plasmid DNA recovered from the cells (34). The latter comparison assumes that all of the plasmid DNA isolated from the cells is nuclear and available for replication and does not take into account possible contamination with extracellular plasmid DNA or plasmid DNA that is in an intracellular compartment separate from the nucleus. To solve these problems and enable the identification of genetic elements that are essential for repli-cator function or augment the activity of essential elements, we adopted a quantitative, internally controlled, short-term repli-cation assay which compares the replirepli-cation efficiency of a test plasmid to that of a previously characterized reference plasmid that is replicated once per S phase (19). Using this assay, we characterized the minimal sequences that are required for ini-tiation of DNA replication from within oriP and identified additional elements that are required to ensure that replication will initiate once per cellular S phase.
Multiple functional elements of the oriP replicator. Our analysis of pHEBo-1.1 derivatives bearing mutations in EBNA-1 sites that inhibit binding in vitro showed that all four EBNA-1 binding sites within oriP DS are required for full replicator activity in Raji cells. These findings provide an explanation for the conservation of all four EBNA-1 binding sites in EBV type 1 and type 2 isolates. Although we have not conducted quan-titative analyses of the replication efficiencies of these plasmids in other EBNA-1-expressing cells, the reduced copy number of these plasmids in the D98/Raji hybrid cell line suggests that the requirement for all four sites is not restricted to B lymphocytes (10). Yates et al. reported thatoriPplasmids bearing six-base substitutions near the center of each of the EBNA-1 binding sites in DS transformed Raji cells to drug resistance less effi-ciently than a plasmid containing four intact sites. In addition, the copy numbers of these mutated plasmids were reduced in an EBNA-1-expressing human osteosarcoma cell line as well as in Raji cells (46). Because the mutations created for their experiments may have altered genetic elements in addition to the EBNA-1 binding sites, it was not certain that the effects of the mutations were due to the disruption of the EBNA-1 bind-ing sites. The data presented here substantiate previous results and support two conclusions: all four EBNA-1 binding sites are required for full replicator activity, and this requirement is likely universal and not cell type specific.
Despite the fact that all four EBNA-1 binding sites within
oriP DS are required for full replicator activity, significant activity was observed when both sites within either pair were inactivated by point mutations or one pair was deleted (10, 46; this study). Thus, only one pair of EBNA-1 binding sites is required for minimal replicator activity. The similar transfor-mation frequencies and copy numbers observed with plasmids
on November 9, 2019 by guest
http://jvi.asm.org/
bearing mutations within either pair of EBNA-1 binding sites in previous studies led to the conclusion that the paired sites contribute similarly to replicator activity (10, 46). However, data presented here clearly show that the paired EBNA-1 binding sites are not functionally equivalent. A plasmid bearing inactivating mutations within binding sites 1 and 2 replicated once every two cellular S phases, on average, while a plasmid with inactivating mutations within binding sites 3 and 4 repli-cated less than once every three S phases. Because EBNA-1 binding sites 3 and 4 contribute more to replicator activity than sites 1 and 2, we consider sites 3 and 4 to be components of the minimal replicator element and sites 1 and 2 to be components of an ancillary element.
Previous studies placed the right-hand boundary of theoriP
replicator between EBNA-1 binding sites 1 and 2 and con-cluded that the sequence to the right of binding site 2 (EBV nt 9107 to 9516) is dispensable for replicator function (17, 32, 44). Notably, the copy numbers oforiPplasmids that lacked either nt 9107 to 9516 or 9135 to 9516 were reduced in comparison to that of a plasmid that contained this sequence, suggesting that an auxiliary element(s) is contained within this DNA fragment (32, 44). The results of experiments presented here showed that the deletion of nt 9138 to 9516 reduced replication effi-ciency approximately 30% per cell generation and that this effect was most likely due to the elimination of an element(s) that is required for full replicator activity. The deletion of nt 9138 to 9516 did not impinge on nonamercand the 2 bp to its right, which preserved a purine and pyrimidine match with the repeats flanking EBNA-1 sites 3 and 4 (Fig. 2). Therefore, we conclude that the loss of replicator activity was due to the elimination of a functional element other than nonamerc.
Kirchmaier and Sugden identified an element that supports inefficient DNA replication in the absence oforiPDS (termed Rep*) within EBV nt 9364 to 9667. Plasmids bearingoriPFR and three copies of Rep* replicated more efficiently than a plasmid bearing only one copy (17). pHEBo-1.1⌬9138-9459 and pHEBo-1.1⌬9138-9516 lack 95 and 152 bp, respectively, of the DNA fragment harboring Rep*, and it is possible that the reduced replication efficiencies of these plasmids are due to the disruption of this functional element. However, Rep* was not found to improve the efficiency of replication of anoriP
plasmid containing EBV nt 8995 to 9137 (17), suggesting that the auxiliary element(s) to the right of nonamer c may be distinct from Rep*. Additional experiments are necessary to define this element(s) and determine how it facilitates oriP
plasmid DNA replication. Further work is also required to determine if another functional element of theoriPreplicator is located to the left of nonamera, as was suggested by the large difference in replication efficiencies of pHEBo-2.2.5 and pHEBo-2.2.20 (Fig. 6).
The minimal replicator.EBNA-1 binding sites 3 and 4 did not support the initiation of DNA replication by themselves, but minimal replicator activity was observed when both of the 14-bp repeats that flank these EBNA-1 binding sites were also present. It is unlikely that this effect is due to an alteration in the position of the EBNA-1 binding sites relative to flanking sequences because the presence of only repeataor repeat b
adjacent to EBNA-1 binding sites 3 and 4 did not provide for detectable activity (Fig. 6) and because substitution mutations within the 14-bp repeats reduced plasmid copy number when
EBNA-1 binding site 1 or sites 1 and 2 were deleted (46). Because the EBNA-1 binding sites are essential components of theoriP replicator (10) and because sites 3 and 4 contribute more to replicator activity than sites 1 and 2, we conclude that the minimal replicator is contained within the 65-bp fragment encompassing sites 3 and 4 and the flanking 14-bp repeats. Yates et al. showed that substitution mutations that substan-tially altered all three of the nonamer repeats had no apparent effect on plasmid maintenance. This result led them to con-clude that the nonamer repeats are not part of the minimal replicator. These same mutations had a profound effect, how-ever, when they were introduced intooriPplasmids that con-tained only “one-half” of the DS. Based on these and other results, they proposed that the minimal replicator of oriP is composed of two EBNA-1 binding sites with 21-bp center-to-center spacing (46). Although it is not clear why the mutation of all three nonamer repeats was found to have no apparent effect when assayed in the context of an otherwise wild-type replicator, results presented here demonstrate that the EBNA-1 binding sites do not provide for significant replicator activity in isolation.
EBNA-1 is bound to its recognition elements within the DS throughout the cell cycle, indicating that a cell cycle-regulated event distinct from the binding of EBNA-1 to the replicator is responsible for the regulated replication oforiPplasmids (14, 26). Results from in vivo dimethyl sulfate footprinting per-formed with synchronized Raji cells provided evidence for the cell cycle-regulated interaction of a protein with the nonamer repeats. Specifically, increased protection or reactivity of cer-tain guanines within each of the nonamer repeats was observed in cells blocked in late G1, and this pattern of protection and
reactivity differed from that observed in cells blocked in mitosis (26). These changes in methylation protection or reactivity sug-gest that a protein is bound to the nonamer repeats in late G1
but not in mitosis. This pattern is reminiscent of cell cycle-de-pendent alterations in the DNase I footprints at a yeast chro-mosomal origin of DNA replication that have been attributed to the formation and dissociation of the prereplication com-plex (5). The relative replication efficiencies of plasmids bear-ing substitution mutations within individual or paired EBNA-1 binding sites or 5-bp insertion mutations between paired sites strongly support the conclusion that protein-protein interac-tions occur not only between EBNA-1 dimers bound to the paired sites but also between EBNA-1 and an unidentified host cell protein(s) bound to the flanking repeats. These findings lead us to propose a model for the initiation of DNA replica-tion atoriP in which EBNA-1 plays a crucial role by the re-cruitment of a host cell protein(s) to three sites in the DS.
ACKNOWLEDGMENTS
We thank K. Takada for providing Akata cells and L. Moore and W. Bauer for assistance with statistical analysis. We also thank A. Sten-lund for many helpful discussions and M. Turner and P. Hearing for critical review of the manuscript.
This work was supported by grants from the National Cancer Insti-tute (CA75992 and 5T32 CA09176).
REFERENCES
1.Adams, A.1987. Replication of latent Epstein-Barr virus genomes in Raji cells. J. Virol.61:1743–1746.
2.Ambinder, R. F., W. A. Shah, D. R. Rawlins, G. S. Hayward, and S. D. Hayward.1990. Definition of the sequence requirements for binding of the