Vol. 41, No. 3 JOURNALOFVIROLOGY, Mar. 1982,p.%5-973
0022-538X/82/030965-09$02.00/0
UV
Irradiation Analysis of Complementation Between,
and
Replication of,
RNA-Negative
Temperature-Sensitive
Mutants
of Newcastle Disease Virus
MARK E. PEEPLES AND MICHAEL A. BRATT*
Departmentof Molecular Genetics andMicrobiology, UniversityofMassachusetts MedicalSchool,
Worcester,Massachusetts01605
Received 12 August1981/Accepted2November1981
Random UV irradiation-induced lesions destroy the infectivity of Newcastle
disease virus (NDV)by blocking downstreamtranscriptionfrom the single viral
promoter.Thenucleocapsid-associated polypeptidesmostlikelytobe involved in
RNA synthesis are located at the extreme ends of the genome: NP and P are
promoterproximal genes, and L is the most distal gene.Weattempted to order the
twotemperature-sensitive (ts)RNA-negative (RNA-) mutant groups ofNDVby
determining the UV target sizes for the complementing abilities ofmutants Aland
El. After UV irradiation, El was unable to complementAl,aresult compatible
with the A mutation lying in the L gene. In contrast, after UVirradiation, Alwas
able tocomplement El for both virusproductionand viralprotein synthesis,with
a target size most consistent with the E mutation lying in the P gene.
UV-irradiated virus was unable toreplicateasindicatedbyits absencein theyieldsof
multiply infectedcells, eitherasinfectiousvirus or asparticleswith complement-ingactivity. After irradiation,ts mutantBlAP,withanon-tsmutationaffectingthe electrophoretic mobility ofthe Pprotein, complementedEl ina mannersimilarto
Al, but it did not amplify the expression of AP in infected cells. This too is
consistent with irradiated virusbeingunable toreplicate despitethepresenceof
thecomponents needed forreplication ofEl. AthighUVdoses, Al wasable to
complement El in adifferent, UV-resistant manner,probably bydirect donation
of input polypeptides. Multiplicity reactivation haspreviously been observedat
high-multiplicity infection by UV-irradiated paramyxoviruses. Inthis case,
viri-ons which are noninfectious because they lack a protein component may be
activatedby a protein from irradiated virions.
Asaparamyxovirus, Newcastle disease virus
(NDV) employsthenegative-strandvirus
strate-gy. Thefirstvirus-specific syntheticeventinan
infected cell is the production ofmRNA (plus strand). With thetranslation ofthese mRNA's, replication (thesynthesis of genome-sized
plus-strand RNA and then completenegative-strand
RNA)begins. Transcriptionfrom these progeny
genomes amplifies viral mRNAs and
conse-quentlyviralproteins.
Genetic analysis of NDV ishampered bythe
peculiarities of negative-strand RNA viruses, which contain a completely covalently linked
genome (reviewed by Bratt and Hightower[4]).
Recombination has not yet been demonstrated (11, 19, 21). If recombination did exist, it might easilybe obscured by the high rate of mutation (42, 24) or by the tendency of NDV to form multiploid particles containing complementing genomes (11, 20, 23, 28).
UV irradiation has been usedas aneffective
tool to circumvent some of these problems.
IrTadiation ofNDV inactivates infectivity with single-hit kinetics (5, 7, 32, 39) and blocks
mRNA transcription (7). Recent irradiation
studies have shown that the NDV genes are sequentially transcribed from a single promoter (9), as previously shown for Sendai virus (18) and vesicular stomatitis virus (VSV) (1, 2). Genes for the three proteins associated with viral nucleocapsids (6, 10, 40), and thought to be
involved in NDV-specific RNA synthesis,
ap-pearwidely separated on the genome; thosefor the NP and P proteins are least sensitive to irradiation and thus closest to the promoter, whereas that for the L protein is as sensitive as infectivityand thus furthest from the promoter
(9).
Two ofthe five complementation groups of
NDVtemperature-sensitive (ts) mutants isolated
by Tsipisand Bratt (42), groups A and E, have
defects affecting RIN4A synthesis (42; M. E.
Peeples, L. L. Rasenas, and M. A. Bratt,
sub-mitted forpublication). As yet, no definite
as-965
on November 10, 2019 by guest
http://jvi.asm.org/
signment of viral proteins to these groups has
been made. We postulatedthat the UV
sensitiv-ity ofaparticular ts mutant's ability to
comple-ment another unirradiated ts mutant should be
determined by the distance between the required gene and the promoter. If some or all of the
NDV genes fromUV-irradiated virus were able
to complement NDV ts mutants, the relative
order of these mutant genes might be
deter-mined. UV irradiation has successfully been
used to order amber mutants of phage T4 (25)
and RNA-negative (RNA-) ts mutants of
Sind-bis virus(17). Surviving genes of UV-irradiated
VSV have been shown to complement group II
andIV mutants (14, 15).
UV-inactivated NDV at high multiplicity can
produce greater than predicted levels of infected cellsorvirus (16, 29).Thismultiplicity
reactiva-tion could be due to cooperation among, or
repair of, damaged virus or, alternatively, to a
nongeneticphenomenon involving the donation ofarequired protein(s)from damaged virus to
anotherwisepotentiallyinfectious viruslacking
thatprotein(4).
We found that UV-irradiation-damaged
ge-nomes of NDV were unabletoreplicate.
How-ever,irradiated Al wasstill abletocomplement
El, but irradiated El was no longer able to
complement Al. Targetsizes were comparable
to those of the P protein (E gene) and the L
protein (A gene). At high radiation doses, Al
was able to complement El in aUV-resistant,
multiplicity of infection (MOI)-dependent
man-ner. This complementation probably reflects
protein transfer from input irradiated virions and provides apossible explanation formultiplicity reactivation.
MATERIALSAND METHODS
Cellcultures. Primaryand secondary chicken
em-bryo cell cultures were maintained in the standard
medium described by Hightower and Bratt (26) at
39.5°C in a 5% CO2 atmosphere. For yield
experi-ments,secondarycultureswereused 24 h afterplating
in 35-mm tissue culture dishes. Forplaquetitrations,
secondary cultures were used as they reached
con-fluency (24 to 48 h after plating) in 60-mm tissue
culture dishes.
Virus stocks. Wild-type virus, AV-WT, was
previ-ouslyclonedfrom the Australia Victoria(1932)strain
of NDV(3). AV-WTwastheparntfor the seriesofts
mutantsisolatedbyTsipisand Bratt(42).Themajorts
mutantsused in thisreportwereAl andEl,
comple-menting mutants with defects in RNAsynthesis(42).
Another tsmutant,BlAP, spontaneouslyaroseduring
recloning of mutant Bl. Bl hasadefect in thegene for
the HNprotein (38). BlAPdiffers fromBl inhavinga
second, non-tsmutation which alters themigrationof
proteinPwithoutalteringitsplatingefficiency,
virus-specific RNAsynthesis,orabilitytocomplementAl
andEl.
Virus stocks weregrown in the allantoicsacof
10-day-oldembryonatedheneggs at 36°C.Allantoicfluid,
harvested after the death of themajority of embryos
(48 to 64h), was concentrated, purified as described
previously (8, 43), and stored at -70°C.
Plaque assays.Titration of infectivity was performed
asdescribedpreviously (3). Plates were incubated at
permissive temperature (37.5°C).
Complementation experiments. Confluentcell
mono-layerswereinoculated with virus at 4°C, washed, and
incubatedwithmedia at a nonpermissivetemperature of 41.8°C(42).
The various multiplicities used are described in the
figurelegends. The 9.5-h yield of each virus alone(Ys)
was subtracted from the yield ofmultipleinfections
(YM).The resultsare presented as the ratio of the YM
- Ys values ofcrosses of complementing mutants,
where one parent was treated with UV irradiation
beforeinfection, to YM- Ysof asimilar cross, where
neither parent was irradiated.
UV irradiation. Two milliliters of virus diluted in standard buffer (0.01 M Tris, pH 7.4, 0.1 N NaCl, and
0.002 M EDTA) wasplaced in an uncovered 60-mm
tissue culture dish and constantly agitated during
exposure to UV irradiation from a SylvaniaG15T8
germicidallamp at adistance of 78 cm.
Quantification of infected cell proteins. After the
medium was removed from infected cultures at 9.5 h, the cultures were washed with Hanks balanced salts
solution(HBSS; GIBCO Laboratories)and labeled for
30 min with 20 ,uCiof[35S]methionine per ml(New
EnglandNuclear Corp.) as described byHightoweret
al. (27). Cultures were washed with HBSS andlysedin
gelsample buffer (0.125 MTris-hydrochloride,pH 6.8,
20%o glycerol, 10%o 2-mercaptoethanol, 6% sodium
dodecylsulfate[SDS],and0.0001%bromphenol blue). Equal portions were electrophoresed at a constant 30
mA on10%polyacrylamidegels(SDS-PAGE)by the
method of Laemmli (30). The gels were dried and
exposed to Kodak Royal X-Omat film. The
autoradio-grams were scanned with anOrtekdensitometer, and
the areas underpeaks were calculated with a Wang
digitizer.
RESULTS
Complementation between A and E mutants.
Mutants of group A complement mutant El at
nonpermissive temperature, producing 50 to
2,000 times greater yields than the sum of the
single-infection yields (42). At permissive
tem-perature, Al makes small plaques
distinguish-able fromEl'sAV-WT-likelargeplaques.Using
this phenotype facilitated the identification of
theprogeny of eachmutantin mixedinfections.
Theyields of crosses between Al and El always
contained both parents(Table 1);thus,
comple-mentation functions in both directions.
UV irradiation of onepartnerof Al xEl. UV
irradiation inactivatedtheinfectivity ofAl,El,
and AV-WT with identical single-hit kinetics (Fig. 1A).
To determine the complementing ability of
UV-irradiated AlorEl,cellswereinfected with
the irradiatedparentat anMOI of1
(preirradia-tion) and with the nonirradiated parent at an
MOI of5.Virtuallyallofthecellswereinfected
on November 10, 2019 by guest
http://jvi.asm.org/
UV IRRADIATION OF NDV ts MUTANTS %7
TABLE 1. Relative yields ofAlandElin complementary crosses
Al and Elyielda
MOI(El) 0 1 5
(AI) % %
PFU/ml Total PFU/ml TotalPFU/ml
El Al El Al
0 1.5 x 101 4.1 x 102
1 6.0 x 101 2.2 x 105 46 54 4.5x 105 78 22
5 3.0 x 102 2.2 x 105 46 54 3.6 x 105 70 30
a Yields from these infections were titrated for infectivity at 37.5°C.
Al
makespredominantly small plaquesandEl makes predominantly large plaques.
with the nonirradiated virus. In addition, many
were infected with a single virion from the
irradiated parent. This procedure was used in a
series of Al xEl crosses in which either Al or
Elhad been subjected toincreasingdosesofUV
irradiation. The nonpermissive temperature yieldsfrom such a mixedinfectionareplotted in
Fig.1B. Inthe series of crosses in whichElwas
the irradiated parent (Al x Eluv),the curvefor
yield reduction closely followed the loss of El
0 z z
I-1
z
6
k-input infectivity (Fig. 1A);the target size for the
A gene, which El must provide for Al, was
apparently the same asthetarget size for
infec-tivity. However, in El x Aluv, Al's abilityto
complement El appeared much more resistant
to UV irradiation thanitsinfectivity; thetarget
sizefor theEgene, which Al mustprovide El,
was much smaller (approximately four times)
than that forinfectivity.
AV-WTcould besubstitutedfor either
irradi-MINUTES UV MINUTES UV
FIG. 1. UVsensitivityofinfectivity and complementing ability. (A) Infectivity remaining after irradiation of
AV-WT (*), Al (+), and El(x).(B)Yields fromAl (MOI=5)xEl (MOI=l)uv(0) and from El (MOI=5)x
Al (MOI = l)uv (v). Inthis and all subsequent figures, the yields from single infections (Ys) have been
subtracted from theyieldsofmultipleinfections(YM)butareincluded (As; Es)ontheleft side of thefigure (when
they fall within thefigure) togive anidea ofbackground levels. WhenASorES values do not fall within the
figure, theyaredesignatedAS4 orESI.
VOL. 41,1982
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.491.51.438.335.618.2]Z EX 0
U_
A5 O
0.1 02 0.3 04 05 0.6
MINUTES U V
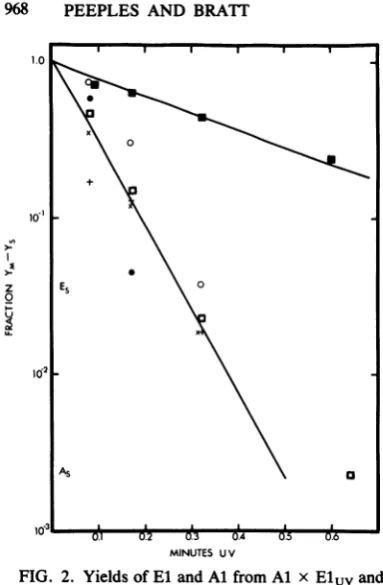
FIG. 2. Yields ofEland Al fromAl x Eluvand
El x Aluv.Nonirradiatedparent,MOI =5;
irradiat-edparent,MOI=1.Large (El) and small(Al) plaque
components of yieldswere counted. El (O) and Al
(0) plaque componentsof Al x ElUVyields;El (v)
andAl(0) plaquecomponentsof El x Aluvyields;
infectivity remaining afterirradiation ofAl (+)orEl
(x). Fraction ofeachmutantin nonirradiated Al xEl
yieldis1.0.
ated Al or irradiated El in these crosses (not
shown).Thedifficultywithusing AV-WT is that
high single-infection yields at low UV doses
must be subtracted from the mixed-infection
yields. Thetsmutantsdidnothavethisproblem
since they produced little background yield at
nonpermissivetemperature.
Relative yields ofAl and El. The yields from
Al x Eluv and El x Aluv could be further
analyzed by plaque size to determine which
mutantwasproduced undertheseconditions. In
Fig. 2, the Al and El componentsof the yields
arecompared.In the Al x Eluv yield, bothAl
and El declined atsimilarrates. Thisindicates
thatonly those Al-infected cells coinfected with
surviving infectious El were able to produce
virus. In contrast, the El component ofEl x
Aluvyielddecreased ataratesimilar to thatof
thetotalyield (Fig. 1B),whereas theyield ofAl
declinedataratesimilartothatofitsremaining
input infectivity, indicating that it was not
re-pairedtoaninfectiousstate atadetectablerate
eventhough itwasable tocomplement.
Analysisof El x Aluvyield for Al-type
com-plementing activity. Infectious Al quickly
disap-peared from the yield of El x Aluv with
increasing irradiation. In this circumstance
Aluv might conceivably be able to replicate,
producing noninfectious virions. These
nonin-fectious Al virions might then beable to
com-plement Elashad theparentalirradiated Al and
would thuspotentially be detected by this
com-plementing activity. Cellswere coinfectedwith
yields from El X Aluv and either Al or El.
Within the El x Aluvyield, the relative
repre-sentation of El complementing ability
in-creased, whereas therepresentationof Al
com-plementing ability decreased,atarate similarto
thatof theinactivationofinput infectivity (Table
2). Therefore, the only Al-type complementing
activity present in these yields was the yield
from the remaining infectious Al.
Viral protein synthesis in infected ceUs. To
determine whethertherelativeyieldsfrom Al x
Eluv- and El x Aluv-infected cells reflected
cellularevents, viralprotein synthesiswas
ana-lyzed in these cells. An experiment similar to
that inFig. 1Bwasperformed (Fig. 3A). These
samecellswerelabeled with [35S]methionine for
30 min immediately after removal of the yield
media. Thecelllysateswere displayed by
SDS-PAGE,andviralpolypeptidesNPplusPand M
were quantified from the autoradiograms (Fig.
3B). The rate of inactivation of virus-specific
protein synthesis was similar to the rate of
inactivation of yield, indicating
complementa-tionatthesynthetic level.
Relative contribution of gene products from
irradiated and nonirradiated parents. Since Al
and El polypeptides have identical migration
rates onSDS-PAGE, the experiment shown in
Fig. 3 provided information on total viral
pro-teins synthesized in these cells, but not the
relativecontributions of each parent.However,
BlAP, a recloned ts mutant Bl with a non-ts
[image:4.491.55.247.51.344.2]mutationaffectingthemigrationof the Pprotein
TABLE 2.
Al-type
complementationactivityin El xAluv yieldsComplementedwithb:
Yieldfroma: Al El
PFU/ml % PFU/ml %
Elx Alo 1.3x 103 100 7.8 x 102 100
El x A10.32C 2.7 x 103 207 1.8 x 102 23
El x Al064d 2.4 x 103 184 10 1.2
aInput = 8.8 x 103PFU; MOI = 0.01.
bMOI= 1.
c Yieldfrom
El
xAluv whereAl
wasUVirradiat-edfor0.32min.
dYieldfrom
El
x AluvwhereAl
was UVirradiat-ed for0.64min.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.491.263.454.535.670.2]UV IRRADIATION OF NDV ts MUTANTS 969
>)
E5+ \
0.3
'SI 0
u s
0~~~~~~~
0
0
Minutes UV
FIG. 3. Comparative effects ofUV irradiation of
one parent on yields and viral protein synthesis in cultures multiply infected with Al and El. (A) UV
inactivationof Al(+)andEl (x)infectivity. Yield of
Al(MOI =5) x El (MOI= 1)uv(0) and yield of El
(MOI = 5) x Al (MOI = 1)uv (0). (B) Relative
amountofviral polypeptidesNP+ Pand M made in
thesamecells in the 30-minperiod after removal of the
yield medium at 9.5 h postinfection. Proteins were
quantified from electropherogramsof polyacrylamide
gelsasdescribed in Materialsand Methods. From Al
X Eluv: NP +P(O),M(O).From El xAluv:NP+
P(*),M(E).
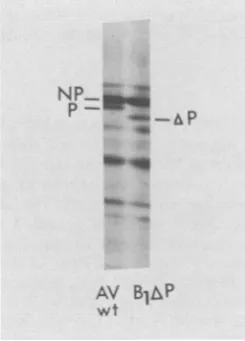
(Fig. 4),wasused astheirradiatedparentinan
experimentlike that shown inFig.3. Justas
AV-WT couldsubstitute for the irradiated parent, so too could BlAP, with the advantage that the inactivation rate ofone protein, AP, could be
measured. BlAP's abilityto complement either
Al or El (Fig. 5A) was similar to that of the
irradiated parents in Fig. 3A. Viral proteins
synthesizedin Al x BlAPuv-infectedcellswere
inactivated at the same rate as Al x Eluv
proteins or infectivity. However, in El x
BlAPuv-infected cells, viral protein synthesis
declined ataratesimilarto that of El x Aluv
with the exception of AP. The synthesis of AP
was inactivated at a rate similar to the rate of
inactivation ofinfectivity. Since proteins labeled
under theseconditions represent products from
amplified transcripts (products from primary
transcripts are not detectable under the low-MOI conditions used here; M. E. Peeples and
M. A.Bratt,unpublished data), these results are
consistent with the AP being produced only from spared, infectious BlAP genomes. The UV inac-tivation rate of replication (and subsequent sec-ondary transcription resulting in protein
synthe-sis)appeared to be similar to that of infectivity.
By thiscriterion, UV-irradiated BlAP is unable
to replicate and amplify its transcription and
translation, even though all the replication
ma-chinery necessary to replicate El, and therefore presumably BlAP, is present.
Complementation targetsize. Table 3presents
Do
values, the amount of UV irradiationre-quired to reduce activity to37% survival,
calcu-lated frominactivation curves similar to those in
Fig. 1B.Thesevalues are comparedwith those
previously calculated byCollinset al. (9)for the
gene targetsizes. The target size of
complement-ingability in Al x Eluv was similar to that of
infectivity and the L gene. The target size of
complementing ability in El xAluvwassimilar
to that of the P gene. These results are most
consistentwithAl providingthePgeneproduct
toEl and are compatible with El providingthe
Lgene product to Al.
Effects of high UV dose on yield from El x
Aluv. Inactivation of the ability of Aluv to
complementElappeared to bequitelinearover
the dose range examined (up to 1.3 min). To
P=
p
m..
AV B1AP
wt
FIG. 4. Migrational difference in P protein from
mutant BlAP. Autoradiogram of10%oSDS-PAGE of AV-WT- andBlAP-infected cells labeled for 30 min
with [35S]methionine (20 ,Ci/ml) at 9.5 h
postinfec-tion. VOL.41, 1982
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.491.47.244.53.400.2] [image:5.491.288.411.445.616.2]° 003
0.1
lAlS
B
.302
0-C
.0
0.
Minutes UV
Fig. 5. Yield and viralprotein synthesis in cultures
multiply infectedwitheitherAlorEl(MOI= 5)and
BlAP (MOI = 1)uv. (A) UV inactivation of BlAP
infectivity (t). Yield ofAl x BlAPuv (0) and El x
BlAP(0). (B) Relativeamountof viralproteinsasin
Fig.5.From Al xBlPuv: NP+P(O),M(O).From
El X BlAPuv: NP+ P(*), M(U), and AP (A).
determine whether this linear relationship
con-tinuedathigher dosesof UV radiation,Al was
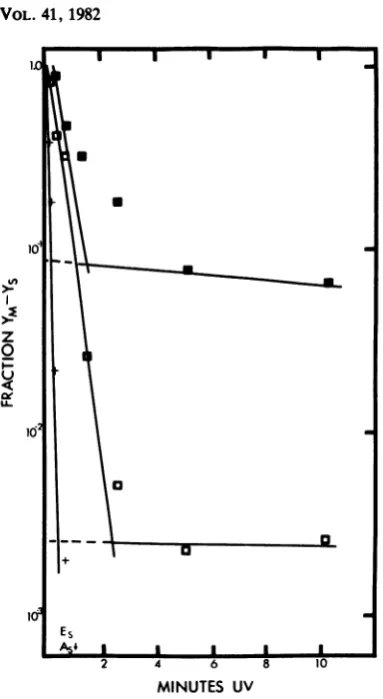
irradiatedupto10.2min,and theyieldof El x Aluv was examined. The inactivation of Al's
complementing ability became resistant (the
slope decreased) between 3 and 5 min and
remained resistantthrough10min (Fig. 6). This
resistance, as Deutsch and Brun(14)have
sug-gestedfrom their VSVstudies,representsa
UV-resistanttarget,possiblyaprotein.If this istrue,
increasing the MOI of Aluv will increase the
input proteinandconsequentlyincrease the
lev-el of the UV-resistantplateau.InFig. 6, Aluvat
anMOI of25 resulted inaplateaulevel
approxi-mately 30-fold higherthananMOI of 1. It thus
appears that the protein required for El could dissociate fromAluv and associate withEl.
DISCUSSION
Replication. Evenin the presence of
comple-menting genomes, UV-irradiated genomes do not appear to be replicated (or repaired): (i) with increasing UV irradiation, the representation of infectious Al or Al-type complementing activity
in El x
Aluv
yields decreases at the same rateas Al input infectivity; and (ii) The amount of
the AP markerprotein synthesized from
ampli-fied genomes in El x
BlAPuv
infected cellsdecreases with UV irradiation at the same rate
as infectivity, whereas the other viral proteins
decrease at a ratesimilar to the yield rate. A UV
lesion appears to inactivate the ability of the
BlAP genome to replicate and amplify viral
protein synthesis and particle production even thoughallof the requirementsfor replicationare
present,since El does replicate. (This would not
be the casewith irradiated AV-WT alone or a ts
mutantalone at permissivetemperature, since a
UV lesion would prevent complete transcrip-tion. As a result, the genome would not be suppliedwith all of the polypeptidesrequired for replication.)Therefore, not only istranscription blocked byUVirradiation(7, 9),it is nowclear
that replication is also blocked by UV
irradia-tion.
A gene. Several pieces of evidence suggest
that the A gene codes for the L protein. (i) The L
gene is the largest NDV gene and by target
theory should represent the largest group of
mutants, which it does (42). (ii) The
noncyto-pathic (nc) mutants of NDV, which are uniform-ly deficient in RNA synthesis and accumulation
of the L protein in infected cells (33), will
complement El, but not group A mutants, for RNA synthesis (34). Thus, the nc mutants
ap-pear to share defects in the L polypeptide and
the A gene. (iii) The results of the
UV-comple-mentation experiments described here are also consistent with the A gene coding for the L protein. The UV target size for the A gene is the entire genome, similar to the conclusion of
Col-TABLE 3. Comparison of target sizes for
complementationwith targetsizes for genes
Determination (erg/MM2)'DO Determinationetri (erg/mmDO 2)b
Infectivity 91 ± 7.6 Infectivity 91
A1x Eluv 104 5.8 L 91
HN 156
M 267
F 351
El xAluv 416 87 P 429
NP 585
aAverageDOofinfectivityassumedtobe thesame
asthat of Collinsetal. (9). Averagesoffiveto seven
experiments.
bFromCollinsetal. (9).
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.491.53.246.52.395.2] [image:6.491.258.448.520.668.2]UV IRRADIATION OF NDV ts MUTANTS 971
z
0
+
Es
2 4 6 8 10
[image:7.491.50.242.51.401.2]MINUTES UV
FIG. 6. High-dose UV irradiation and high-MOI
effects on complementation. Al was irradiated at
various dosesupto 10.2min and usedtoinfect,ata
multiplicity of 1 or 25, cultures which were also
infected with El. El (MOI = 5) x Al (MOI = l)uv
(0); El (MOI=5)xAl(MOI=25)uv (v).Infectivity
of Al remainingafterirradiationis alsoplotted(+).
linsetal.(9)concerningthe Lgene.However,it
is possible that an entire genome target size
might reflect a requirement for replication
in-stead ofaspecific gene.
Egene.After low-doseirradiation,bothAluv
andBlAPuvwereabletomakeenough of the E
gene product to complement the
RNA-synthe-sizing step(s) of transcription orreplication (or both) of El. Their UV target size was most
similar to that of the P gene. Because of the
multiplestepsinvolved incomplementation,this
targetsizemaynotbeexact.Thegeneswiththe
next-larger and next-smaller target sizes are F
andNP,respectively(9). It isunlikelythat the E
genewould codefor the Fprotein, since F isnot
involved in RNAsynthesis. However,NPmust
beconsidered,since both P and NPareprobably
neededinRNA synthesis.
Afterhigh-doseirradiation,Aluvstill
comple-ments El in a UV-resistant,
MOI-dependent
manner.(Thereverseisnot true:
Eluv
is unableto complement Al in a UV-resistant way at a
similar
MOI;
Peeples and Bratt,unpublished
data.)
The E gene product in the inputAluv
virions isprobably this UV-resistant targetand
must,therefore,be abletodissociate from
Aluv
inordertocomplementEl.The NP
polypeptide
is veryfirmlyassociated withthe viralgenomic RNA, an unlikely candidate for a diffusible
agent. In
fact,
NP istheonlyviruspolypeptide
notremovedfromnucleocapsidsbythehighsalt
orin aCsCl gradient
(37).
In addition, the Egene product must be re-quiredinverysmall amounts,perhaps enzymati-cally, since products from single genes (low-doseirradiation)andproteins from input virions (high-doseirradiation) provide enoughfor com-plementation. NP is required in large
stoichio-metric amountstocovereach50Sgenome-sized RNAasit isproducedin the cell.Requirements
for Parenot asclear,but much less P than NP is
foundinnucleocapsids, and the number ofthose
which are functional or necessary is unclear.
Furthermore, Deutschetal.(15) found thatonly
group II VSV mutants could be
complemented
by both a functional surviving gene and by a
structuralproteinofUV-irradiated virus,similar
totheEl mutant ofNDVdescribed here.Group
II mutants of VSVrepresent lesions in the NS
gene (31, 35). Both the P polypeptide of NDV
and theNSpolypeptideofVSV are
phosphory-lated minornucleocapsid-associatedspecies(36,
40, 41), andboth genes arepenultimatetotheir
promoters,just after theNPandN gene,
respec-tively (1, 2, 9). It seems more likely, then, that
the E gene of NDV represents the P protein,
ratherthanthe NPprotein.
Multiplicity reactivation.Complementationby
alow MOI ofirradiated virus was due onlyto
theinfectious virions presentbeforeirradiation,
since both theAl X
Eluv
and the El xAluv
curves were straight linesintercepting the
ordi-nate at1.0. IncreasingtheMOIoftheirradiated
virusto5resulted in an apparentmultihitcurve
with an extrapolated ordinate intercept of
ap-proximately 5 (datanotshown). Thisreflectsthe
initial infection of an average five infectious
virions per celland the survival ofatleast one
virion per cell at low doses of irradiation. In
complementation experimentsin which theMOI
of one parent was progressively decreased be-low 1 by dilution instead ofirradiation, yields
decreasedwithsingle-hit kinetics. Thedecrease
precisely mimicked theUVinactivation of
infec-tivity, regardless of which parent was diluted
(data not presented). The single-hit kinetics
again imply that asingle infectious virion, and
only a single infectious virion, is required for
complementation.
VOL.41, 1982
on November 10, 2019 by guest
http://jvi.asm.org/
Drake (16) and Kirvaitis and Simon (29) have demonstrated that high-MOI infections with
UV-irradiated NDV result in more infected cells
or a greater yield than expected from surviving infectivity. Recombination cannot explain this phenomenon, since it has not been found for paramyxoviruses (reviewed by Bratt and High-tower [4]). It is also unlikely that two viruses lethally irradiated in different parts of their genome could complement each other, produc-ing complementproduc-ing heterozygotes, since NDV is a single transcription unit: a lesion anywhere would destroy downstream genes (9). Potential-ly infectious but non-plaque-forming virus was
detected by Granoff (20, 22). We have shown
here that input virions can supply a UV-resistant function, probably a protein, possibly the P protein, resulting in complementation of an NDV mutant. This process was especially obvi-ous when a high MOI of highly inactivated virus was used. It has previously been demonstrated that VSV proteins from high MOIs of highly UV-irradiated virus can complement ts mutants (12, 13, 15). These findings support the Bratt and
Hightower (4) hypothesis: a lethally irradiated
virus might be able to provide a necessary function to a virion which is noninfectious due to defective protein packaging rather than a defec-tive genome. If this were the case, the
inactiva-tion of infectivity might be a deceptively low
estimate of the remaining potentially infectious virus.
ACKNOWLEDGMENTS
We thank Rhona Glickman, Judith Brackett, Michael Glass, and Timothy Biliouris for their excellent technicalassistance, Chris Biron, RonIorio,Larry Hightower, Chuck Madansky, and RayWelsh for helpful discussions, and Susan Longwell,
AnneChojnicki,and Judith Brackett for their help in
prepara-tion of this manuscript.
We are grateful to the National Institute of Allergy and Infectious Diseases, Public Health Service, for the grant (AI12467) that supported this project and the fellowship (AI05874) that supported M.E.P.
LITERATURECITED
1. Abraham, G., and A. K. Banerjee. 1976. Sequential tran-scription of the genes of vesicularstomatitis virus. Proc. Natl. Acad. Sci. U.S.A. 73:1504-1508.
2. Ball, L. A., and C. N.White. 1976. Order of transcription of genes of vesicular stomatitis virus. Proc. Natl. Acad. Sci. U.S.A. 73:442-446.
3. Bratt, M. A., and W. R. Gallaher. 1969. Preliminary analysis of the requirements forfusion from within and fusion from without by Newcastle disease virus. Proc. Natl. Acad. Sci. U.S.A.64:536-543.
4. Bratt, M. A., and L. E.Hightower. 1977. Genetics and paragenetic phenomena of paramyxoviruses, p. 457-533. In H. Fraenkel-Conrat and R. R.Wagner (ed.), Compre-hensive virology, vol. 9. PlenumPublishingCorp., New York.
5. Bratt, M. A., and H. Rubin. 1968. Specificinterference among strains of Newcastle diseasevirus. II.Comparison of interference by active and inactive virus. Virology 35:381-394.
6. Chambers, P., and A. C. R. Sampson. 1980. A new structural protein for Newcastle disease virus. J. Gen. Virol.50:155-166.
7. Clavell, L. A., and M. A. Bratt. 1971. Relationship be-tweenribonucleic acid-synthesizingcapacity of ultravio-let-irradiated Newcastle disease virus andits ability to induceinterferon,J.Virol.8:500-508.
8. Clavell,L.A.,andM. A. Bratt.1972. Hemolytic interac-tionof Newcastle disease virus and chickenerythrocytes. II. Determining factors.Appl. Microbiol. 23:461-470. 9. Collins, P. L., L. E. Hightower, and L. A. Ball. 1980.
Transcriptional mapfor Newcastle disease virus. J.Virol. 35:682-693.
10. Colonno, R. J., and H. 0. Stone. 1976. Isolation ofa transcriptive complex from Newcastle diseasevirions. J. Virol. 19:1080-1089.
11. Dahlberg, J. E., and E. H. Simon.1969.Recombination in Newcastledisease virus(NDV): theproblem of comple-menting heterozygotes. Virology 38:490-493.
12. Deutsch, V. 1975. Nongenetic complementationof group V temperature-sensitive mutants of vesicular stomatitis virus by UV-irradiated virus. J. Virol. 15:798-805. 13. Deutsch, V. 1976. ParentalG protein reincorporationbya
vesicular stomatitis virustemperature-sensitive mutant of complementation group V atnonpermissivetemperature. Virology 69:607-616.
14. Deutsch, V., and G. Brun. 1978. Rescue atnonpermissive temperature of complementation group II temperature-sensitive mutants of vesicular stomatitis virus by UV-irradiated VSV.Virology 87:96-108.
15. Deutsch, V., B. Muel, and G.Brun. 1977. Action spectra for the rescue of temperature-sensitivemutants of vesicu-lar stomatitis virus by ultraviolet-irradiated virions at nonpermissive temperature.Virology 77:294-305. 16. Drake, J. W.1962.Multiplicityreactivation ofNewcastle
disease virus. J. Bacteriol. 84:352-356.
17. Fuller, F. J., and P. I. Marcus. 1980. Sindbis virus. I. Gene order of translation in vivo.Virology 107:441-451. 18. Glazier, K., R. Raghow, and D. W. Kingsbury. 1977.
Regulation of Sendai virus transcription: evidence for a single promoter in vivo. J. Virol. 21:863-871.
19. Granoff, A. 1959. Studies on mixedinfection with New-castle disease virus. I. Isolation of Newcastle disease virus mutants and tests for genetic recombination be-tween them. Virology 9:636-648.
20. Granoff, A. 1959. Studies on mixedinfection with New-castle disease virus. II. The occurrence of Newcastle disease virus heterozygotes and the study of phenotypic mixing involving serotypes andthermalstability. Virology 9:649-670.
21. Granoff, A. 1961. Induction ofNewcastle disease virus mutants with nitrous acid. Virology 13:402-408. 22. Granoff, A. 1961. Studies on mixed infection with
New-castle disease virus. III. Activation ofnonplaque-forming virus by plaque forming virus. Virology14:143-144. 23. Granoff, A. 1962. Heterozygosis andphenotypic mixing
with Newcastle disease virus. Cold SpringHarbor Symp. Quant. Biol. 27:319-326.
24. Granoff, A. 1964. Nature of Newcastle disease virus population, p. 107-118. In R. P.Hanson(ed.), Newcastle disease virus, an evolving pathogen. The University of Wisconsin Press, Madison.
25. Hercules, K., and W.Sauerbler.1973.Transcription units in bacteriophage T4. J. Virol. 12:872-881.
26. Hightower, L. E., andM. A.Bratt. 1974. Protein synthesis in Newcastle diseasevirus-infected chicken embryo cells. J. Virol. 13:788-800.
27.Hlightower,L. E., T. G.Morrison, and M. A.Bratt. 1975. Relationshipsamong the polypeptides ofNewcastle dis-ease virus. J. Virol. 16:1599-1607.
28. Kingsbury,D. W.,and A.Granoff. 1970. Studies on mixed infection with Newcastledisease virus. IV. On the struc-ture of heterozygotes. Virology42:262-265.
29. Kirvaitis, J., and E. H. Simon. 1965. A radiobiological study of the development ofNewcastle disease virus.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
UV IRRADIATION OF NDV ts MUTANTS 973 Virology 26:545-553.
30. Laemmli, U. K. 1970. Cleavage of structural proteins during assembly of the head ofbacteriophage T4. Nature (London) 277:680-685.
31. LaFay,F., andJ.Benejean.1981.Temperaturesensitive mutantsof vesicular stomatitisvirus: tryptic peptidemaps
ofthe protein modified incomplementationgroupIIand IV.Virology 111:93-102.
32. Levinson, W., and H. Rubin. 1966. Radiation studies of aviantumorviruses and Newcastle disease virus.
Virolo-gy28:533-542.
33. Madansky,C.H., and M. A. Bratt. 1978.Noncytopathic mutants of Newcastle disease virus: comparison with naturally occurringavirulent strains,p.709-720.InB. W.
J. Mahy and R. D. Barry (ed.),Negative strand viruses and the host cell. Academic Press, Inc., London. 34. Madansky, C. H., and M. A. Bratt. 1981.Noncytopathic
mutantsof Newcastledisease virusaredefective in
virus-specific RNAsynthesis. J. Virol. 37:317-327.
35. Metzel, P. S., and M. E. Relchmann. 1981. Characteriza-tion ofvesicular stomatitis virusmutantsbypartial prote-olysis. J. Virol. 37:248-255.
36. Morrison,T.G., and D. Simpson.1980.Synthesis, stabil-ity and cleavage ofNewcastle disease virus glycoproteins intheabsence of glycosylation. J. Virol. 36:171-180.
37. Mountcastle,W.E.,R. W.Compans,L. A.Caliguiri,and P. W. Choppin. 1970. Nucleocapsidproteinsubunits of simian virus5,Newcastlediseasevirus,andSendai virus. J.Virol. 6:677-684.
38. Peeples,M.E., J.P.Gallagher, andM. A. Bratt. 1981.
Permissive temperatureanalysis ofRNA' temperature-sensitivemutantsof Newcastlediseasevirus,p.567-572.
In D. H. L. Bishop and R. W. Compans (ed.), The replication of negative strand viruses. Elsevier Press, NewYork.
39. Roman, J. A., and E. H. Simon. 1976. Morphologic heterogeneityinegg-andmonolayer-propagated Newcas-tlediseasevirus.Virology69:287-297.
40. Smith,G.W.,andL. E.Hightower.1981.Identificationof
the Pproteinsand otherdisulfide-linkedand phosphory-lated proteins of Newcastle disease virus. J. Virol. 37:256-267.
41. Sokol, F.,and H. F.Clark. 1973.Phosphoproteins, struc-tural componentsofrhabdoviruses.Virology52:246-263. 42. TsipIs, J.E.,andM. A. Bratt.1976.Isolation and
prelimi-narycharacterizationof temperature-sensitivemutantsof Newcastledisease virus. J. Virol. 18:848-855.
43. Weiss,S. R., and M. A Bratt. 1974. Polyadenylate se-quencesonNewcastlediseasevirus mRNAsynthesized
invivo and invitro.J. Virol.13:1220-1230. VOL. 41,1982
on November 10, 2019 by guest
http://jvi.asm.org/