0022-538X/07/$08.00⫹0 doi:10.1128/JVI.01601-07
Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Induction of Epidermal Growth Factor Receptor Expression by
Epstein-Barr Virus Latent Membrane Protein 1
C-Terminal-Activating Region 1 Is Mediated by
NF-
B p50 Homodimer/Bcl-3 Complexes
䌤
Natalie J. Thornburg
1and Nancy Raab-Traub
1,2*
Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599,1and
Department of Microbiology-Immunology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 275992
Received 23 July 2007/Accepted 12 September 2007
The Epstein-Barr virus (EBV) is associated with the development of numerous malignancies, including the epithelial malignancy nasopharyngeal carcinoma (NPC). The viral oncoprotein latent membrane protein 1 (LMP1) is expressed in almost all EBV-associated malignancies and has profound effects on gene expression. LMP1 acts as a constitutively active tumor necrosis factor receptor and activates multiple forms of the NF-B family of transcription factors. LMP1 has two domains that both activate NF-B. In epithelial cells, LMP1 C-terminal activating region 1 (CTAR1) uniquely activates p50/p50-, p50/p52-, and p65-containing complexes while CTAR2 activates canonical p50/p65 complexes. CTAR1 also uniquely upregulates the epidermal growth factor receptor (EGFR). In NPC, NF-B p50/p50 homodimers and the transactivator Bcl-3 were detected on the EGFR promoter. In this study, the role of NF-B p50 and Bcl-3 in LMP1-mediated upregulation of EGFR was analyzed. In LMP1-CTAR1-expressing cells, chromatin immunoprecipitation detected p50 and Bcl-3 on the NF-B consensus sites within the egfrpromoter. Transient overexpression of p50 and Bcl-3 increased EGFR expression, confirming the regulation of EGFR by these factors. Treatment with p105/p50 siRNA effectively reduced p105/p50 levels but unexpectedly increased Bcl-3 expression and levels of p50/Bcl-3 com-plexes, resulting in increased EGFR expression. These data suggest that induction of p50/p50/Bcl-3 complexes by LMP1 CTAR1 mediates LMP1-induced EGFR upregulation and that formation of the p50/p50/Bcl-3 complex is negatively regulated by the p105 precursor. The distinct forms of NF-B that are induced by LMP1 CTAR1 likely activate distinct cellular genes.
The Epstein-Barr virus (EBV) is a ubiquitous human her-pesvirus that infects more than 90% of the world’s population and is associated with the development of numerous malignan-cies, such as nasopharyngeal carcinoma (NPC) (12). Latent membrane protein 1 (LMP1) is expressed in most EBV-asso-ciated malignancies, is essential for EBV-induced B-lympho-cyte transformation, and is the EBV oncogene (11). LMP1 induces focus formation in rodent fibroblasts, supports anchor-age-independent growth of cells in soft agar, and supports tumor formation in nude mice (12). LMP1 is an integral mem-brane protein that acts as a constitutively active tumor necrosis factor receptor. The C-terminal domain has two signaling re-gions, CTAR1 and CTAR2, which constitutively associate with tumor necrosis factor receptor-associated factors (TRAFs) (24). Through its association with TRAFs, LMP1 initiates sig-naling events including activation of the NF-B signaling cas-cade (10, 14). The transcriptional up-regulation of multiple cellular genes, such asicam-1, cd80, cd23, cd54, bcl-2, traf1,
a20, andegfr, is mediated by LMP1, and many of these genes are known to be regulated by NF-B (14, 15, 22, 28, 32). In epithelial cells, LMP1 activates at least three distinct types of NF-B complexes (27).
NF-B is a family of transcription factors that regulate a broad range of biological processes, including inflammation, angiogenesis, cell cycle regulation, apoptosis, and oncogenesis (7, 19). There are five mammalian NF-B family members, p50, p52, p65 (RelA), c-Rel, and RelB. The NF-B family members dimerize and bind NF-B consensus sequences in cellular and viral promoters through their Rel homology do-main. The p65, c-Rel, and RelB family members have trans-activation domains that recruit transcriptional machinery to promoters. The activation of NF-B family members is tightly regulated through interactions with inhibitors of NF-B (IB), which sequester NF-B members in the cytosol. Extracellular stimuli, such as binding of tumor necrosis factor to its receptor, induce a kinase cascade that ultimately results in phosphory-lation, ubiquitination, and degradation of an IB, leading to the release and nuclear translocation of bound NF-B. The mammalian IBs include p105 (NFB1, the p50 precursor), p100 (NFB2, the p52 precursor), IB␣, IB, IB␥, IBε, and Bcl-3.
In epithelial cells, CTAR1 activates at least three different dimeric forms of NF-B, including p50/p50 homodimers, p50/ p52 heterodimers, and complexes containing p65 (27). In con-trast, CTAR2 induces only one complex that contains p65. The distinct forms of NF-B induced by CTAR1 or CTAR2 are in part mediated by different signaling pathways. Both CTAR1 and CTAR2 can activate NF-B through the canonical path-way. This pathway is activated through the trimeric IB kinase
* Corresponding author. Mailing address: Lineberger Comprehen-sive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599. Phone: (919) 966-1701. Fax: (919) 966-9673. E-mail: [email protected].
䌤Published ahead of print on 19 September 2007.
12954
on November 8, 2019 by guest
http://jvi.asm.org/
has transactivation domains; however, both can bind the onco-protein Bcl-3 (2, 26). Bcl-3 is an unusual member of the IB family in that it is most commonly expressed in the nucleus and it has a transactivation domain that can provide transactivating function to p50- or p52-containing complexes (2, 26).
Previous examination of xenografted NPC tumors detected p50/p50 homodimers and Bcl-3 by chromatin immunoprecipi-tation (ChIP) on the NF-B sites within theegfrpromoter. In this study, the unique ability of CTAR1 to activate p50/p50 homodimers and upregulate the EGFR was examined. The data indicate that the induction of p50/p50 homodimers by CTAR1 and the increased formation of complexes containing Bcl-3 induce EGFR expression.
MATERIALS AND METHODS
Cell culture and reagents.C33A cervical carcinoma cells were cultured in Dulbecco’s modified Eagle medium (Gibco) supplemented with 10% fetal bo-vine serum (Sigma) and antibiotics at 37°C with 5% CO2.Cells were transfected
using the Fugene 6 transfection reagent (Roche) as directed by the manufac-turer. Stable cell lines were made by transfecting the pCDNA3 vector control or Myc-tagged LMP1 vectors into C33A cells. Forty-eight hours posttransfection, cells were trypsinized, replated, and selected with 0.6 mg/ml G418-supplemented medium. Stable cell lines were passaged in the presence of G418.
Cell extracts and Western blots.Cells were scrape harvested, washed once with cold phosphate-buffered saline (PBS), and lysed with RIPA buffer (10 mM Tris-HCl [pH 8.0], 140 mM NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate [SDS], 1% deoxycholic acid) supplemented with protease and phosphatase inhibitor cocktails (Sigma). Equal amounts of protein were used for SDS-poly-acrylamide gel electrophoresis (PAGE) and Western blotting. Primary antibod-ies used for Western blots include anti-p105/p50, anti--actin, anti-Bcl-3, anti-c-Myc, anti-IKK(Santa Cruz), anti-p65 (Rockland), and anti-p100/p52 (Upstate). A rabbit antiserium raised against the carboxy-terminal 100 amino acids of the EGFR fused to glutathioneS-transferase (kindly provided by H. Shelton Earp) was used to detect EGFR. Secondary antibodies used were horseradish peroxi-dase-conjugated antimouse and antirabbit (Amersham Pharmacia) and antigoat (DAKO). Blots were developed using the Pierce Supersignal West Pico chemi-luminescence system.
Immunoprecipitations.Cells were scraped, washed with cold 1⫻PBS, and lysed in IP buffer (1% Triton X-100, 0.5% of Nonidet P-40, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA) supplemented with protease inhibitor cocktail (Sigma). Seventy-five micrograms of cellular extracts were precleared with GammaBind Plus Sepharose (Amersham Pharmacia) for 2 h at 4°C. Precleared lysates were immunoprecipitated with 1g anti-p105/p50, anti-Bcl-3, anti-p100/p52, or iso-type control antibody overnight. Immunoprecipitations were incubated with GammaBind Plus Sepharose for 2 h at 4°C, washed two times, resuspended in SDS-PAGE sample buffer, boiled, and used for SDS-PAGE and Western blots.
ChIP.ChIPs were performed as previously described (30). Briefly, 1⫻107
cells were scraped, resuspended in 50 ml DMEM, and cross-linked in 1% form-aldehyde for 15 min at room temperature, followed by quenching with 120 mM glycine. The cell pellet was washed with 1⫻PBS and lysed in RIPA buffer (10 mM Tris-HCl [pH 8.0], 140 mM NaCl, 1% Triton X-100, 0.1% SDS, 1%
deoxy-tails [Sigma]) for 15 min on ice, followed by addition of Nonidet P-40 to a final concentration of 1%. Nuclei were pelleted by low-speed centrifugation at 1,200 rpm for 10 min at 4°C. The nuclei were purified using the Optiprep reagent (Sigma) as directed by the manufacturer, as previously described (30). Nuclei were lysed with nuclear extraction buffer (20 mM Tris [pH 8.0], 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, protease inhibitor cocktail
[Sigma], and phosphatase inhibitor cocktail [Sigma]) with the salt concentration adjusted to 400 mM with 5 M NaCl. Insoluble nuclear material was pelleted at high speed for 10 min. EMSAs were performed as previously described (3). An oligonucleotide (UV21) of the NF-B site from the H-2Kb
gene (CA GGGCTGGGGATTCCCCATCTCCCACAGTTTCACTTC) was labeled with [␣-32
P]dCTP using the Klenow fragment from DNA polymerase I. Five micro-grams nuclear extracts were incubated with radiolabeled probe. For supershift assays, 1g antibody was incubated with extracts. Antibodies used were p105/p50, Rel-B, cRel (Santa Cruz), p65 (Rockland), and anti-p100/p52 (Upstate).
Quantitative reverse transcription-PCR.Total RNA was isolated from cells, using an RNeasy minikit as directed by the manufacturer (QIAGEN). Total RNA was quantified by spectrophometric measurements. Primer pairs were designed using the Primer Express program (Applied Biosystems). The actin primer set was 5⬘TCACCCACACTGTGCCCATCTACGA 3⬘and 5⬘CAGCG GAACCGCTCATTGCCAATGG 3⬘. The EGFR primer set was 5⬘TGCGTC TCTTGCCGGAAT 3⬘and 5⬘GGCTCACCCTCCAGAAGC 3⬘. Quantitative reverse transcription-PCR was performed on DNase-treated RNA using the Quantitect SYBR Green RT-PCR kit (QIAGEN) according to the manufactur-er’s directions. Amplification of target sequences was detected using an ABI 7900HT sequence detection system (Applied Biosystems) and analyzed using SDS 2.0 software (Applied Biosystems). The cycle threshold (CT) was
deter-mined as the number of PCR cycles required for a given reaction to reach an arbitrary fluorescence value within the linear amplification range. The change in
CT(⌬CT) was determined between the same gene primer sets and different
samples, and the change in⌬CT(⌬⌬CT) was determined by adjusting for the
difference in the number of cycles required for actin to reach theCT. Since each
PCR cycle results in a twofold amplification of each product, then-fold differ-ence was determined as 2⌬⌬CT. Each sample was analyzed in triplicate, and the
standard error was determined.
siRNA.Chemically synthesized small interfering RNA (siRNA) pools were purchased from Dharmacon, targeting an irrelevant RNA, NF-B1, and Bcl-3. Either 100 or 50 pmol siRNA was transfected into stable cells using a Lipo-fectamine 2000 transfection reagent (Invitrogen) as directed by the manufac-turer. Cells were harvested approximately 36 h posttransfection and were used for Western blot analysis, quantitative reverse transcription-PCR, or immuno-precipitations.
RESULTS
NF-B p50 binds Bcl-3 and immunoprecipitates with the EGFR promoter.Previous studies revealed that LMP1 upregu-lates the EGFR at the mRNA level (22). In EBV-positive NPC tumors, NF-B p50 homodimers and Bcl-3 could be immuno-precipitated with NF-B consensus sites in theegfrpromoter (30). In order to determine if LMP1 can induce the binding of p50 and Bcl-3 to theegfrpromoter, C33A cells stably
on November 8, 2019 by guest
ing the pCDNA3 vector control, full-length LMP1, LMP1(1-231) (LMP1 that retains CTAR1 but not CTAR2; hereafter referred to as 1-231), and LMP1(⌬187-351) (LMP1 that re-tains CTAR2 but not CTAR1; hereafter referred to as⌬ 187-351) were examined by ChIP using primers that flank the NF-B sites in theegfrpromoter. In C33A stable cells, LMP1 expression and EGFR upregulation were confirmed by immu-noblotting (Fig. 1A). As described previously, LMP1 activated EGFR through CTAR1. The deletion mutant that retains CTAR1, 1-231, strongly increased EGFR, while the deletion mutant that retains CTAR2, ⌬187-351, did not upregulate EGFR (Fig. 1A). By ChIP, precipitation with anti-p105/p50 weakly detected the NF-B sites in theegfrpromoter in cells expressing LMP1 and 1-231 (Fig. 1B, lanes 9 and 10) but not in pCDNA3 cells or ⌬187-351 cells (Fig. 1B, lanes 8 and 11). Bcl-3 was detected on the egfrpromoter in 1-231-expressing cells (Fig. 1B, lane 14) and weakly detected on theegfr pro-moter in LMP1-expressing cells (Fig. 1B, lane 13). Bcl-3 was not detected on theegfrpromoter in pCDNA3- or⌬ 187-351-expressing cells (Fig. 1B, lanes 12 and 15). This ChIP corre-lated with EGFR expression. In LMP1- and 1-231-expressing cells, EGFR was upregulated and p50 and Bcl-3 were detected on theegfrpromoter. A very low level of EGFR was detected in vector control cells and⌬187-351-expressing cells, and Bcl-3 and p50 were not detected on theegfr promoter. The other NF-B family members activated by LMP1, p65, p52, and RelB were not detected on theegfrpromoter (Fig. 1B, lanes 23 to 34). The absence of p52 on theegfrpromoter indicates that the unique activation of the noncanonical pathway by CTAR1 does not activate EGFR expression and that EGFR expression is specifically correlated with the presence of p50 and Bcl-3 on theegfrpromoter.
Transient expression of p50 and Bcl-3 increase EGFR.In order to determine if expression of p50 and/or Bcl-3 is suffi-cient to mediate an increase in EGFR protein levels, C33A cells were transiently transfected with vector control, p50,
[image:3.585.138.449.69.229.2]Bcl-3, or p50 and Bcl-3. Expression of each was confirmed by immunoblotting, and equal loading was confirmed with a -ac-tin immunoblot (Fig. 2). EGFR expression levels were also detected by immunoblotting. Transient expression of p50, Bcl-3, or both detectably increased EGFR compared to results with the vector control (Fig. 2). The intensities of the EGFR bands shown in Fig. 2 were quantified using the Image J 1.32j computer program and were listed immediately above the cor-responding bands. This experiment has been repeated five times; however, the values were calculated from the represen-tative Western blot shown in Fig. 2. Cells expressing p50 alone had an approximately twofold increase in the EGFR protein. Cells expressing Bcl-3 alone had an approximately threefold increase in the EGFR protein. Cells expressing both p50 and Bcl-3 had an approximately fivefold increase in the EGFR
FIG. 1. NF-B p50 and Bcl-3 immunoprecipitate on the EGFR promoter. (A) EGFR and LMP1 expression was examined by immunoblotting with C33A cells stably expressing Myc-tagged LMP1 or deletion mutant 1-231 or⌬187-351 with anti-c-mycand anti-EGFR. The bottom panel is a loading control. (B) A chromatin immunoprecipitation of p50, Bcl-3, p100/p52, p65, and RelB on theegfrpromoter was performed with C33A cells stably expressing LMP1 or deletion mutant 1-231 or⌬187-351. Lanes 1 and 16 are DNA markers, lanes 2 and 17 are water PCR controls, lanes 3 and 18 are genomic DNA from C33A cells, lanes 4 to 7 are immunoprecipitated in the absence of antibody, lanes 8 to 11 are immunoprecipitated with anti-p105/p50 (␣-p105/p50), lanes 12 to 15 are immunoprecipitated with anti-Bcl-3 (␣-Bcl-3), lanes 19 to 22 are input DNA from all four stable cell lines, lanes 23 to 26 are immunoprecipitated with anti-p100/p52 (␣-p100/p52), lanes 27 to 30 are immunoprecipitated with anti-p65 (␣-p65), and lanes 31 to 34 are immunoprecipitated with anti-RelB (␣-RelB).
FIG. 2. Transient expression of p50 and Bcl-3 increases EGFR protein. C33A cells were transiently transfected with p50 and Bcl-3, and levels of EGFR were examined by immunoblotting. Equal loading was confirmed with anti--actin. EGFR levels were measured by den-sitometry of the immunoblot in the top panel. The Image J 1.32j program was used to calculate the intensity of each band from one Western blot. Pixel densities are listed above their corresponding bands. The blot is representative of five independent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:3.585.360.479.519.650.2]protein. These data indicate that both p50 and Bcl-3 contribute to upregulation of EGFR.
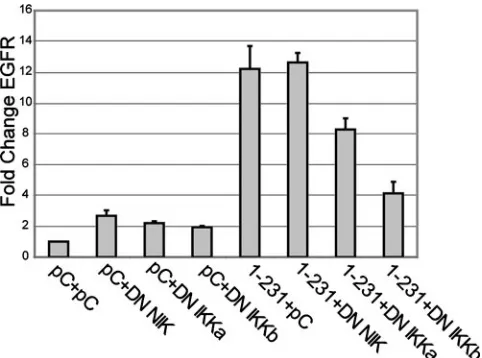
Increased EGFR expression requires canonical NF-B ac-tivation. Several studies have determined that CTAR2 acti-vates NF-B via the canonical pathway that is dependent upon the trimeric IKK complex composed of IKK␣, IKK, and IKK␥(NEMO). CTAR1 activates the NF-B canonical path-way and the noncanonical pathpath-way that induces processing of p100 to p52. This processing is dependent on NIK and IKK␣ but is independent of the trimeric complex (1, 5, 29). A third pathway has been studied, using knockout murine embryo fi-broblasts, that was IKK dependent and considered atypical canonical (17). To identify the pathways that contribute to CTAR1-induced EGFR expression, dominant-negative forms (DN) of NIK, IKK␣, and IKKwere transiently expressed in cells stably expressing 1-231. EGFR expression was analyzed by quantitative reverse transcription-PCR. Expression of DN NIK, IKK␣, and IKK did not significantly affect the trace levels of EGFR in vector control cells (Fig. 3). In cells express-ing CTAR1 (1-231), DN NIK also did not significantly changes EGFR levels. DN IKK␣and DN IKKdecreased EGFR ex-pression in 1-231 cells, with the most dramatic decrease in-duced by DN IKK. The inhibition of EGFR up-regulation by both DN IKK␣and DN IKKsuggests that CTAR1-mediated effects on EGFR are regulated in part by the canonical path-way. This is consistent with previously published data that demonstrated that expression of a DN IB␣ also inhibited LMP1-induced EGFR expression (21). The lack of an effect by the DN NIK indicates that the induction of EGFR by CTAR1 is not due to the noncanonical pathway.
[image:4.585.43.284.68.247.2]DN IKK decreases nuclear p50 and EGFR protein. To further characterize the forms of NF-B that contribute to LMP1-mediated EGFR up-regulation, cells stably expressing the pCDNA3 vector control, LMP1, 1-231, and⌬187-351 were transiently transfected with pCDNA3 vector control, wild-type (wt) IKK, or the IKKDN double point mutant (SS 177, 181 AA) (DN IKKSSAA). Expression of LMP1, deletion mu-tants, and IKK was confirmed by immunoblotting (Fig. 4, middle two panels). Equal loading was assessed by Western blotting (Fig. 4, bottom panel). NF-B activation was detected by EMSA with a radiolabeled oligonucleotide probe (UV21) with the NF-B consensus site from the MHC class I promoter, using nuclear extracts (Fig. 4, top panel). EGFR levels were measured by immunoblotting (Fig. 4, second panel). In C33A epithelial cells, LMP1 and 1-231 activate p50 homodimers, p50/p52 heterodimers, and p52/p65 heterodimers and ⌬ 187-351 activates p52/p65 heterodimers (27). Transfection of wt IKK and DN IKK into pCDNA3 control cells did not change NF-B complex levels (Fig. 4, lanes 2 to 4). In both LMP1- and 1-231-expressing cells, three complexes were de-tected (Fig. 4, upper panel, lanes 5 and 8). The identities of these complexes were confirmed by supershift analysis (data not shown). Expression of DN IKKin LMP1 cells reduced all complexes, and that in 1-231 cells almost completely elimi-nated them. In particular, the strong nuclear p50/p50 ho-modimer level was greatly diminished (Fig. 4, upper panel, lanes 7 and 10). This decrease in nuclear p50/p50 homodimer levels correlated with a decrease in EGFR levels (Fig. 4, lanes 7 and 10). In the CTAR2 containing⌬187-351 cells, p52/p65
FIG. 3. Increased EGFR expression requires NF-B activation. Stable cell lines expressing the pCDNA vector control or 1-231 were transiently transfected with the vector control, DN NIK, IKK␣, or IKK. RNA was harvested and used for quantitative reverse transcrip-tion-PCR using primer sets for-actin and EGFR. The graph shows then-fold change from the level for pCDNA3 stable cells transfected with the vector control and represents the average from separate experiments. All samples are normalized to vector control results, and EGFR levels were additionally normalized to the sample’s correspond-ing-actin level. The bars represent then-fold change in EGFR levels from levels for pCDNA3 cells transfected with vector control. Each sample was performed in triplicate, and the standard error is indicated. The graph shown is representative of three independent experiments.
FIG. 4. DN IKKdecreases nuclear p50 homodimers and EGFR protein. C33A cells transiently transfected with pCDNA3 vector con-trol, wild type IKK(WT IKK), or double-point-mutant (SS 177, 181 AA) DN IKK(DN IKK) were tested for nuclear NF-B complexes by EMSA. Levels of EGFR, LMP1, and IKK were examined by immunoblotting. The LMP1 expression panel is a composite from two different gels. Equal loading was confirmed by using a loading control (bottom panel). Lane 1 is a probe-alone control for the EMSA. Lanes 2 to 4 are pCDNA3 stable cells transiently transfected with pCDNA3, wt IKK, or DN IKK; lanes 5 to 7 are LMP1 stable cells transfected with pCDNA3, wt IKK, or DN IKK; lanes 8 to 10 are 1-231 stable cells transfected with pCDNA3, wt IKK, or DN IKK, and lanes 11 to 13 are⌬187-351 stable cells transfected with pCDNA3, wt IKK, or DN IKK. Arrows identify the complexes. The EMSA and Western blots are each representative of four independent experiments.
on November 8, 2019 by guest
[image:4.585.315.523.68.246.2]heterodimers and p50 homodimers were detected (Fig. 4, up-per panel, lanes 2 and 11, upup-per panel). Expression of wt IKK in ⌬187-351 cells greatly increased the amount of the p65-containing complex, indicating activation of the canonical pathway. In addition, the p50/p50 homodimer was increased, but this did not affect the trace levels of EGFR (Fig. 4, lanes 11 and 12, top two panels). Expression of the DN IKKin⌬ 187-351 cells eliminated the p52/65 form but surprisingly increased the p50/50 homodimer. This increase did not increase the level of EGFR. The effects of expression of the wt IKKand DN IKKon the complexes detected by EMSA in CTAR1 suggest that the IKKsubunit contributes to p50/p50 homodimer ac-tivation and LMP1-mediated EGFR up-regulation. The ab-sence of EGFR up-regulation in the⌬187-351 cells by either the wt IKKor DN IKK, both of which increased the levels of p50 homodimers, suggests that p50 homodimers are not sufficient to activate EGFR expression and that an additional activity is required that is not induced by⌬187-351.
NF-B1 precursor negatively regulates p50/50 complexes and EGFR expression.In order to determine if p50 and Bcl-3 are necessary for CTAR1-mediated EGFR upregulation, siRNAs targeting NF-B1 and Bcl-3 were utilized to decrease p50 and Bcl-3 expression. Cells were transfected with siRNA directed against an irrelevant RNA, the p50 precursor, NF-B1 (p105), Bcl-3, or both, and the effect on the cellular proteins was determined by immunoblotting (Fig. 5). Transfection with NF-B1 siRNA effectively decreased expression of the p50 precur-sor protein, p105, and p50 (Fig. 5A, lane 2), and the decrease was dose dependent (Fig. 5A, lanes 2 and 4). In contrast, transfection with Bcl-3 siRNA did not affect the levels of cel-lular Bcl-3 (Fig. 5A, lanes 3 and 4). Interestingly, transfection with NF-B1 siRNA significantly increased the amount of Bcl-3, suggesting that p105 or p50 may negatively regulate Bcl-3. Transfection with NF-B1 siRNA or Bcl-3 siRNA did not alter the other NF-B family member, p65 (Fig. 5A).
The regulation of the processing of p105 to p50 is distinct from that of the processing of p100 to p52. In the pcDNA3 control cells, processed p50 was constitutively abundant while processed p52 was detected at very low levels (Fig. 5B). LMP1 CTAR1 induces processing of p100 to p52 (1, 5, 17, 29). This effect was apparent with greatly increased p52 in the 1-231-expressing cells (Fig. 5B). In contrast, the relative amount of p50/p105 over the pcDNA3 control was slightly increased by LMP1 CTAR1.
[image:5.585.137.448.70.256.2]To determine the effect of NF-B1 siRNA treatment on EGFR upregulation, the siRNAs were transfected into vector control cells or cells expressing the LMP1 mutant, 1-231, which has the strongest activation of EGFR. NF-B p105 and p50 were effectively decreased by siRNA transfection in both pCDNA3- and 1-231-expressing cells (Fig. 5B). The NF-B1 siRNA or Bcl-3 siRNA had a very slight effect on the total amount of p100 and p52 and did not affect the LMP1-induced processing of p100 (Fig. 5B). Surprisingly, siRNA-mediated decrease of NF-B1 either alone or in combination with Bcl-3 increased EGFR expression compared to the irrelevant siRNA control in 1-231-expressing cells (Fig. 5B, lanes 5, 6, and 8). The effects of EGFR expression were quantified using the Image J 1.32j computer program, and pixel intensities were listed immediately above the corresponding bands. This exper-iment was repeated three times; however, the values were calculated from one representative Western blot. The in-creased EGFR protein was not due to a decrease in the NF-B-regulated inhibitory protein IB␣, as levels of IB␣were not affected by the siRNA-mediated decrease of NF-B1 or the increased Bcl-3 (Fig. 5B, bottom panel). However, the increase in EGFR correlated with the increased Bcl-3 in the NF-B1 siRNA-transfected 1-231 cells (Fig. 5A). It is known that the precursors for p52 and p50 contain ankyrin repeats and can function as repressors. The increase in EGFR induced by the siRNA-mediated decrease in p105 likely reflects the
FIG. 5. NF-B1 siRNA negatively regulates p105/p50 expression and increases Bcl-3 and EGFR expression. (A) C33A cells were transfected with 100 pmol irrelevant siRNA, 100 pmol NF-B1 siRNA, 100 pmol Bcl-3 siRNA, or 50 pmol NF-B1 and Bcl-3 siRNAs. Knockdown was confirmed by immunoblotting. (B) C33A cells stably expressing pCDNA3 vector control or 1-231 were transfected with 100 pmol irrelevant siRNA, 100 pmol NF-B1 siRNA, 100 pmol Bcl-3 siRNA, or 50 pmol NF-B1 and Bcl-3 siRNAs. NF-B and EGFR protein levels were examined by immunoblotting. The Image J 1.32j computer program was used to calculate the intensity of each EGFR band from one Western blot. Pixel density values are listed above the corresponding bands. Units are arbitrary. The blot is representative of five independent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
inhibitory properties of p105 for the availability of the p50 homodimer complex.
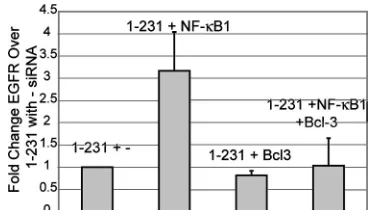
To determine if the increase in EGFR protein levels after siRNA treatment of NF-B1 and Bcl-3 occurred at the RNA level, quantitative reverse transcription-PCR was performed on vector control cells and cells expressing 1-231 transiently transfected with siRNA of irrelevant RNA, NF-B1, and Bcl-3. PCR was performed with actspecific primers for RNA in-tegrity control and EGFR-specific primers. Each sample was normalized to RNA from cells expressing 1-231 transfected with irrelevant siRNA. The graph in Fig. 6 shows then-fold change over the level for 1-231 with irrelevant siRNA and represents results from four independent experiments. The actin levels were unchanged and therefore confirm the integ-rity of the RNA (data not shown). Cells expressing 1-231 and transfected with NF-B1 siRNA had an approximately three-fold increase of EGFR RNA over that for 1-231-expressing cells. Bcl-3 siRNA expression alone or in combination with NF-B1 siRNA did not significantly change EGFR RNA (Fig. 6). These results confirmed the immunoblot analysis and sug-gested that siRNA degradation of cellular NF-B1 increased EGFR protein and RNA levels and that this correlated with the increase in Bcl-3.
Increased Bcl-3 coimmunoprecipitates with NF-B1 after transfection with NF-B1 siRNA.To determine the effect of an siRNA-mediated decrease of NF-B1 and Bcl-3 on forma-tion of the p50/Bcl-3 complex, coimmunoprecipitaforma-tion of p105/ p50, p100/p52, and Bcl-3 was analyzed. P105 and p50 did not immunoprecipitate with anti-p100/p52 (Fig. 7A, lanes 8 to 12). A considerable level of NF-B p50 immunoprecipitated with anti-Bcl-3 in vector control pCDNA3 cells (Fig. 7A, lane 13), and increased p50 immunoprecipitated with anti-Bcl-3 in 1-231 cells treated with irrelevant siRNA (Fig. 7A, lane 14). Al-though the amount of p50 was greatly reduced by transfection with NF-B1 siRNA (Fig. 5A), the amount of p50 that coim-munoprecipitated with Bcl-3 was only slightly affected (Fig. 7A, lanes 15 and 17). The siRNA-mediated decrease of p105/p50 and the consequential increase in Bcl-3 resulted in more Bcl-3
[image:6.585.69.254.68.173.2]for 1-231 stable cells transfected with irrelevant siRNA, and each EGFR PCR was further normalized to the-actin level in its corre-sponding sample. The graph showsn-fold change in EGFR RNA level over that for 1-231 with irrelevant siRNA and is a representative result from five independent experiments.
FIG. 7. Increased Bcl-3 coimmunoprecipitates with NF-B1 after transfection with NF-B1 siRNA. (A) Coimmunoprecipitation of p105/ p50 with p100/p52 and Bcl-3 was measured after knockdown with 100 pmol irrelevant siRNA, 100 pmol NF-B1 siRNA, 100 pmol Bcl-3 siRNA, or 50 pmol NF-B1 and Bcl-3 in C33A cells stably expressing pCDNA3 or 1-231. Stable cells were transfected with the siRNA indicated above the immunoblot. Lanes 1 and 2 are a direct load of protein lysates from pCDNA3 and 1-231 stable cells. Lanes 3 to 7 are immunoprecipitated with rabbit immunoglobulin G (IgG) isotype control, lanes 8 to 12 are immunoprecipitated with anti-p100/p52 (␣-p100/p52), and lanes 13 to 17 are immunoprecipitated with anti-Bcl-3 (␣-Bcl-3). All immunoprecipi-tates were immunoblotted with anti-p50. (B) Coimmunoprecipitation of Bcl-3 with anti-p105/p50 (␣-p105/p50) and anti-p100/p52 was measured after knockdown with siRNAs as described for panel A with C33A cells stably expressing pCDNA3 or 1-231. Stable cells were transfected with the siRNA indicated above the immunoblot. Lanes 1 and 2 are direct loads of protein lysates from pCDNA3 and 1-231 stable cells. Lanes 3 to 7 are immunoprecipitated with goat IgG isotype control, lanes 8 to 12 are immunoprecipitated with anti-p105/p50, and lanes 13 to 17 are immuno-precipitated with anti-p100/p52. The complexes were immunoblotted with anti-Bcl-3. (C) Coimmunoprecipitation of anti-p100/p52 with anti-p105/ p50 and Bcl-3 was measured after siRNA knockdown as described for panel A in pCDNA3 and 1-231 stable C33A cells. Stable cells were transfected with the siRNA indicated above the immunoblot. Lanes 1 and 2 are a direct load of protein lysates from pCDNA3 and 1-231 stable cells. Lanes 3 to 7 are immunoprecipitated with mouse IgG isotype control, lanes 8 to 12 are immunoprecipitated with anti-p105/p50, and lanes 13 to 17 are immunoprecipitated with anti-Bcl-3. The immunoprecipitates were immunoblotted with anti-p52.
on November 8, 2019 by guest
[image:6.585.311.526.73.460.2]in complexes immunoprecipitated with anti-p105/p50 (Fig. 7B, lanes 10 and 12). Immunoprecipitation with anti-p100/p52 con-firmed that in C33A cells expressing 1-231, p100/p52 did not bind p50 (Fig. 7A, lanes 8 to 12) or Bcl-3 (Fig. 7B, lanes 13 to 17). NF-B p100/p52 also did not immunoprecipitate with ei-ther anti-p105/p50 or anti-Bcl-3 (Fig. 7C, lanes 8 to 17). These data indicate that transfection of NF-B1 siRNA did not in-duce formation of p52/Bcl-3 complexes. These results reveal that despite a significant decrease in the overall pool of p105/ p50, the remaining p50 binds more efficiently to Bcl-3 in the absence of p105 and that the increased EGFR expression cor-related with increased complex formation between p50 and Bcl-3.
DISCUSSION
The data presented in this study indicate that the LMP1 CTAR1 domain induces the binding of NF-B p50 and Bcl-3 to the NF-B sites in theegfrpromoter in C33A cells and this correlated with EGFR upregulation (Fig. 1). It has previously been determined thategfrhas five NF-B consensus binding sites in its promoter, and p50 specifically binds to four of those sites (25). Furthermore, it was determined that in xenografted NPCs, both p50 and Bcl-3 coimmunoprecipitate with three of those sites in theegfrpromoter (30). The data in this paper support the in vivo model in which p50 and Bcl-3 are present on theegfrpromoter when CTAR1 is present. Of note, anti-p100/p52, anti-p65, and anti-RelB did not immunoprecipitate the NF-B sites in theegfrpromoter, indicating that in C33A cells, these family members do not promote EGFR tion. The absence of p52 or RelB confirms that the upregula-tion of EGFR by CTAR1 was not mediated by the effects of CTAR1 on the noncanonical NF-B pathway.
Expression of p50 and Bcl-3 in the absence of LMP1 also increased EGFR expression (Fig. 2). NF-B p50/p50 and p52/ p52 homodimers have been thought of as being transcription-ally inhibitory because neither p50 nor p52 has transactivation domains. NF-B p50 can bind the transcriptionally inhibitory histone deacetylase 1 (34). However, if bound to Bcl-3, p50/p50 and p52/p52 homodimers may be transcriptionally active (2, 33). It has been shown that p52/Bcl-3 complexes transcription-ally upregulated cyclin D1 and promoted cell cycle progression more efficiently than other NF-B family members (33). NF-B p50 may also directly activate cellular promoters, and the antiapoptotic protein Bcl-2, which is also upregulated by LMP1, can be transcriptionally regulated by p50 homodimers (13, 28). In EBV-positive samples of NPC, p50/p50 ho-modimers preferentially bound Bcl-3 and did not bind histone deacetylase 1, and both p50 and Bcl-3 were detected by ChIP on the EGFR promoter (30).
NF-B activity is controlled by a kinase cascade that begins with an extracellular signal that leads to activation of the IKK complex and phosphorylation of IB. The IKK complex is a trimeric complex consisting of two catalytic domains, IKK␣ and IKK, and a regulatory domain, IKK␥(NEMO). IKKis the dominant kinase in phosphorylation of IB (7). NIK can also phosphorylate and activate IKK␣, which can phosphory-late the p52 precursor, p100, in an IKK␥-independent manner to produce p52. LMP1 CTAR1 activates NF-B through both IKK␥-dependent and noncanonical IKK␥-independent
path-ways, mediates NF-B release from IB, and induces phosphor-ylation of p100 followed by processing of p100 to p52 (1, 5, 17, 29). In contrast, CTAR2 induces canonical NF-B activation through the IKK␥-dependent IKK complex.
The role of NF-B was confirmed in this study, since trans-fection of DN IKK␣ and IKKdecreased EGFR in cells ex-pressing 1-231 (Fig. 3 and 4). These findings confirm the pre-vious inhibition of LMP1-mediated EGFR upregulation by a DN IB␣ (20). In 1-231 cells, transient transfection of DN IKKdecreased EGFR RNA threefold, while transient trans-fection of DN NIK did not change EGFR and DN IKK␣ decreased EGFR RNA by approximately one-third. The non-canonical processing and activation of p52 by CTAR1 is de-pendent upon NIK and IKK␣; therefore, the minimal effects of DN NIK and DN IKK␣and the more-pronounced inhibition by DN IKKfurther indicate that the CTAR1 upregulation of EGFR is not mediated by p52 and the noncanonical pathway. The data presented here indicate that the unique CTAR1 upregulation of EGFR is linked in part to its induction of p50 homodimers and that IKKis a significant factor in the regu-lation of EGFR by CTAR1.
However, the increased levels of p50 homodimers induced by wt IKKand DN IKKin cells expressing CTAR2-contain-ing⌬187-351 LMP1 were not sufficient to induce EGFR ex-pression. This suggests that other signaling pathways activated by LMP1 CTAR1 contribute to EGFR upregulation in con-junction with NF-B. A recent publication indicated that GSK3 phosphorylates Bcl-3 and mediates its degradation (31). LMP1 activates Akt through CTAR1, and phosphorylated, inactive GSK3is elevated in EBV-infected cells (4, 18). The activation of Akt and inactivation of GSK3 by LMP1 may affect the activity of Bcl-3.
The siRNA-mediated decrease in p105/p50 surprisingly in-creased levels of Bcl-3 and p50/Bcl-3 complexes and correlated with increased levels of EGFR (Fig. 6 and 7). Although the siRNA-mediated decrease in p50 could affect the amount of IB proteins, a change in IB␣ was not detected (Fig. 5B). NF-B p105 is the p50 precursor and is also considered an IB since it can act as an inhibitory molecule by binding and se-questering p50 in the cytoplasm (6, 8, 9, 16). The decrease in the inhibitory p105 likely increases the availability of the re-maining processed p50, which may complex with Bcl-3 more efficiently in the absence of p105 (Fig. 7). Bcl-3 can also inter-act with p52 to form a transcriptionally inter-active complex; how-ever, p52 was not detected on theegfrpromoter in C33A cells (Fig. 1) and p52 was not detected in a complex with p50 or Bcl-3 by coimmunoprecipitation in C33A cells (Fig. 7). These findings support the previous detection of only p50 and Bcl-3 on the egfr promoter in xenografted NPC and indicate that p50/Bcl-3 more likely mediates EGFR upregulation (5, 30). The data presented in this article suggest a new role for NF-B p50/Bcl-3 complexes as transcriptional activators and indicate that this complex likely functions in the transcriptional regu-lation of EGFR.
ACKNOWLEDGMENTS
We gratefully acknowledge H. Shelly Earp for the anti-EGFR rabbit antiserum, Albert S. Baldwin for the IKKexpression construct, and Elliot Kieff for the IKK␣and NIK expression constructs.
This work was supported by NIH grant CA32979 to N.R.-T.
on November 8, 2019 by guest
http://jvi.asm.org/
6.Fan, C. M., and T. Maniatis.1991. Generation of p50 subunit of NF-kappa B by processing of p105 through an ATP-dependent pathway. Nature354:
395–398.
7.Ghosh, S., and M. Karin.2002. Missing pieces in the NF-kappaB puzzle. Cell
109(Suppl.):S81–S96.
8.Hatada, E. N., A. Nieters, F. G. Wulczyn, M. Naumann, R. Meyer, G. Nucifora, T. W. McKeithan, and C. Scheidereit.1992. The ankyrin repeat domains of the NF-kappa B precursor p105 and the protooncogene bcl-3 act as specific inhibitors of NF-kappa B DNA binding. Proc. Natl. Acad. Sci. USA89:2489–2493.
9.Heissmeyer, V., D. Krappmann, F. G. Wulczyn, and C. Scheidereit.1999. NF-kappaB p105 is a target of IkappaB kinases and controls signal induction of Bcl-3-p50 complexes. EMBO J.18:4766–4778.
10.Huen, D. S., S. A. Henderson, D. Croom-Carter, and M. Rowe.1995. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene10:549–560.
11.Kaye, K. M., K. M. Izumi, and E. Kieff.1993. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc. Natl. Acad. Sci. USA90:9150–9154.
12.Kieff, E., and A. B. Rickinson.2001. Epstein-Barr virus and its replication, p. 2511–2573.InD. M. Knipe (ed.), Field’s virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA.
13.Kurland, J. F., R. Kodym, M. D. Story, K. B. Spurgers, T. J. McDonnell, and R. E. Meyn.2001. NF-kappaB1 (p50) homodimers contribute to transcrip-tion of the bcl-2 oncogene. J. Biol. Chem.276:45380–45386.
14.Laherty, C. D., H. M. Hu, A. W. Opipari, F. Wang, and V. M. Dixit.1992. The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor kappa B. J. Biol. Chem.267:24157– 24160.
15.Liebowitz, D., and E. Kieff.1989. Epstein-Barr virus latent membrane pro-tein: induction of B-cell activation antigens and membrane patch formation does not require vimentin. J. Virol.63:4051–4054.
16.Liou, H. C., G. P. Nolan, S. Ghosh, T. Fujita, and D. Baltimore.1992. The NF-kappa B p50 precursor, p105, contains an internal I kappa B-like inhib-itor that preferentially inhibits p50. EMBO J.11:3003–3009.
17.Luftig, M., T. Yasui, V. Soni, M. S. Kang, N. Jacobson, E. Cahir-McFar-land, B. Seed, and E. Kieff.2004. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK alpha-depen-dent noncanonical NF-kappaB activation. Proc. Natl. Acad. Sci. USA
101:141–146.
18.Mainou, B. A., D. N. Everly, Jr., and N. Raab-Traub.2005. Epstein-Barr
24.Mosialos, G., M. Birkenbach, R. Yalamanchili, T. VanArsdale, C. Ware, and E. Kieff.1995. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell80:389– 399.
25.Nishi, H., G. Neta, K. H. Nishi, L. M. Akers, T. Rikiyama, K. N. Proctor, B. A. Murphy, and A. C. Johnson.2003. Analysis of the epidermal growth factor receptor promoter: the effect of nuclear factor-kappaB. Int. J. Mol. Med.11:49–55.
26.Nolan, G. P., T. Fujita, K. Bhatia, C. Huppi, H. C. Liou, M. L. Scott, and D. Baltimore.1993. Thebcl-3proto-oncogene encodes a nuclear IB-like mol-ecule that preferentially interacts with NFB p50 and p52 in a phosphory-lation-dependent manner. Mol. Cell. Biol.13:3557–3566.
27.Paine, E., R. I. Scheinman, A. S. Baldwin, Jr., and N. Raab-Traub.1995. Expression of LMP1 in epithelial cells leads to the activation of a select subset of NF-B/Rel family proteins. J. Virol.69:4572–4576.
28.Rowe, M., M. Peng-Pilon, D. S. Huen, R. Hardy, D. Croom-Carter, E. Lundgren, and A. B. Rickinson.1994. Upregulation ofbcl-2by the Epstein-Barr virus latent membrane protein LMP1: a B-cell-specific response that is delayed relative to NF-B activation and to induction of cell surface markers. J. Virol.68:5602–5612.
29.Saito, N., G. Courtois, A. Chiba, N. Yamamoto, T. Nitta, N. Hironaka, M. Rowe, and S. Yamaoka.2003. Two carboxyl-terminal activation regions of Epstein-Barr virus latent membrane protein 1 activate NF-kappaB through distinct signaling pathways in fibroblast cell lines. J. Biol. Chem.278:46565– 46575.
30.Thornburg, N. J., R. Pathmanathan, and N. Raab-Traub.2003. Activation of nuclear factor-kappaB p50 homodimer/Bcl-3 complexes in nasopharyngeal carcinoma. Cancer Res.63:8293–8301.
31.Viatour, P., E. Dejardin, M. Warnier, F. Lair, E. Claudio, F. Bureau, J. C. Marine, M. P. Merville, U. Maurer, D. Green, J. Piette, U. Siebenlist, V. Bours, and A. Chariot.2004. GSK3-mediated BCL-3 phosphorylation mod-ulates its degradation and its oncogenicity. Mol. Cell16:35–45.
32.Wang, F., C. Gregory, C. Sample, M. Rowe, D. Liebowitz, R. Murray, A. Rickinson, and E. Kieff.1990. Epstein-Barr virus latent membrane protein (LMP1) and nuclear proteins 2 and 3C are effectors of phenotypic changes in B lymphocytes: EBNA-2 and LMP1 cooperatively induce CD23. J. Virol.
64:2309–2318.
33.Westerheide, S. D., M. W. Mayo, V. Anest, J. L. Hanson, and A. S. Baldwin, Jr.2001. The putative oncoprotein Bcl-3 induces cyclin D1 to stimulate G1
transition. Mol. Cell. Biol.21:8428–8436.
34.Zhong, H., M. J. May, E. Jimi, and S. Ghosh.2002. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell9:625–636.