organic papers
o2482
Houet al. C14H14S2 doi:10.1107/S1600536805020908 Acta Cryst.(2005). E61, o2482–o2483
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
1,2-Bis(phenylsulfanyl)ethane
Bao-Hong Hou,aLi-Na Zhou,a Qiu-Xiang Yin,aJing-Kang Wanga and Wei Chena,b*
aThe State Research Center of Industrialization
for Crystallization Technology, Tianjin University, Tianjin 300072, People’s Republic of China, andbTianjin Economic and
Technological Development Area, Tianjin, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.042
wRfactor = 0.131
Data-to-parameter ratio = 19.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography
Printed in Great Britain – all rights reserved
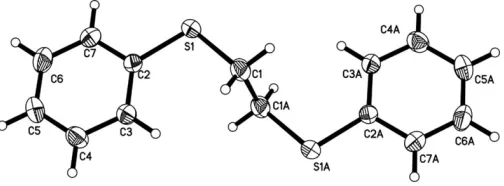
In the title compound, C14H14S2, there is a centre of inversion at the mid-point of the central C—C bond. Excluding H atoms, the molecule adopts an anti conformation, with a planar –S– CH2–CH2–S– spacer unit. The dihedral angle between the phenyl ring and the S—C—C—S chain is 84.52 (18).
Comment
Many complexes of bis(thioether) ligands with transition metal ions, such as silver(I) (Blacket al., 1995), palladium(II) (Errington et al., 1980) and platinum(II) (Murray & Hartley, 1981), have been characterized and have interesting struc-tures. In contrast, the structures of the free ligands have been much less studied. In the present paper, we report the crystal structure of the title compound, a bis(thioether) ligand, viz. 1,2-bis(phenylsulfanyl)ethane (bpte), (I).
The bpte molecule has a centre of inversion at the mid-point of the central C1—C1Abond; symmetry code: (A) 1x,y,
z; Fig. 1]. Excluding H atoms, the molecule adopts an anti conformation (Goodgame et al., 1999), with a planar spacer unit (S1—C1—C1A—S1A). The S—C bond distances and C— S—C angles are comparable to those observed in an analogous compound, 1,4-bis(phenylsulfanyl)butane (Chenet al., 2005). The S1 S1A non-bonded distance is 4.4223 (16) A˚ . The phenyl ring makes a dihedral angle of 84.52 (18) with the plane of the spacer unit (S1—C1—C1A—S1A).
In the crystal structure of the silver(I) nitrate complex with bpte, (II) (Shao et al., 1991), the ligand adopts a different geometry; the orientation of one phenyl group of bpte with respect to the spacer plane is similar to that in the title
[image:1.610.208.460.611.704.2]Received 8 June 2005 Accepted 30 June 2005 Online 9 July 2005
Figure 1
compound, but the other phenyl group is twisted about the S—Cphenyl bond. There is no centre of symmetry and the phenyl rings are inclined to each other by 63.4. A similar
torsion can also be found in the PdCl2complex with bpte, (III) (Wanget al., 1992).
The S—C bond length and S S non-bonded distance in free bpte are slightly shorter than those in complexes (II) and (III), and other analogous bis(thioethers) and corresponding complexes (Buet al., 2002).
Another determination of the title compound is reported in the preceding paper (Awalehet al., 2005).
Experimental
1,2-Bis(phenylsulfanyl)ethane (bpte) was prepared according to a reported procedure (Shao et al., 1991) and the product was char-acterized by NMR. Colourless single crystals of the title compound, suitable for X-ray diffraction, were obtained by slow evaporation at room temperature of a solution in chloroform.1H NMR (CDCl3): 3.02 (t, 4 H), 7.36 (m, 10 H).
Crystal data
C14H14S2
Mr= 246.39
Monoclinic,P21=c
a= 5.8389 (12) A˚
b= 7.6865 (15) A˚
c= 14.124 (3) A˚
= 97.90 (3)
V= 627.9 (2) A˚3
Z= 2
Dx= 1.303 Mg m
3
MoKradiation Cell parameters from 5725
reflections
= 3.0–27.5 = 0.39 mm1
T= 293 (2) K Needle, colourless 0.800.230.10 mm
Data collection
Rigaku R-AXIS RAPID IP area-detector diffractometer
!scans
Absorption correction: multi-scan (ABSCOR; Higashi, 1995)
Tmin= 0.745,Tmax= 0.961
5725 measured reflections
1418 independent reflections 1180 reflections withI> 2(I)
Rint= 0.058 max= 27.5
h=6!7
k=9!9
l=18!18
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.042
wR(F2) = 0.131
S= 1.03 1418 reflections 74 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0735P)2
+ 0.1339P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.48 e A˚ 3
min=0.28 e A˚ 3
Extinction correction:SHELXL97
[image:2.610.312.564.112.147.2]Extinction coefficient: 0.059 (13)
Table 1
Selected geometric parameters (A˚ ,).
S1—C2 1.7609 (17) S1—C1 1.825 (2)
C2—S1—C1 105.15 (9)
All H atoms were positioned geometrically, with Csp2—H = 0.93 A˚ and Csp3—H = 0.97 A˚ ; they were constrained to ride on their parent atoms, withUiso(H) = 1.2Ueq(C).
Data collection:RAPID-AUTO (Rigaku, 2004); cell refinement:
RAPID-AUTO; data reduction:RAPID-AUTO; program(s) used to solve structure: SHELXS97(Sheldrick, 1997); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics:
ORTEPII (Johnson, 1976); software used to prepare material for publication:CrystalStructure(Rigaku, 2004).
We gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 20206022).
References
Awaleh, M. O., Badia, A. & Brisse, F. (2005). Acta Cryst. E61, o2479– o2480.
Black, J. R., Champness, N. R., Levason, W. & Reid, G. (1995).J. Chem. Soc. Chem. Commun.pp. 1277–1278.
Bu, X.-H., Chen, W., Hou, W.-F., Du, M., Zhang, R.-H. & Brisse, F. (2002).
Inorg. Chem.41, 3477–3482.
Chen, W., Hou, B.-H., Zhou, L.-N., Wang, J.-K. & Li H. (2005).Acta Cryst.E61, o1890–o1891.
Errington, J., McDonald, W. S. & Shaw, B. L. (1980).J. Chem. Soc. Dalton Trans.pp. 2309–2315.
Goodgame, D. M. L., Grachvogel, D. A., Hussain, I., White, A. J. P. & Williams, D. J. (1999).Inorg. Chem.38, 2057–2063.
Higashi, T. (1995).ABSCOR. Rigaku Corporation, Tokyo, Japan.
Johnson, C. K. (1976).ORTEPII. Report ORNL-5138. Oak Ridge National Laboratory, Tennessee, USA.
Murray, S. G. & Hartley, F. R. (1981).Chem. Rev.81, 365–414.
Rigaku. (2004). RAPID AUTO andCrystalStructure. Rigaku/MSC Inc., 9009 New Trails Drive, The Woodlands, TX 77381-5209, USA.
Shao, P.-X., Yao, X.-K., Wang, H.-G., Wang, W.-H., Liu, B., Li, M., Luo, L.-W. & Xu D.-H. (1991).Chem. J. Chin. Univ.12, 143–147.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
supporting information
sup-1 Acta Cryst. (2005). E61, o2482–o2483
supporting information
Acta Cryst. (2005). E61, o2482–o2483 [https://doi.org/10.1107/S1600536805020908]
1,2-Bis(phenylsulfanyl)ethane
Bao-Hong Hou, Li-Na Zhou, Qiu-Xiang Yin, Jing-Kang Wang and Wei Chen
1,2-bis(phenylsulfanyl)ethane
Crystal data
C14H14S2 Mr = 246.39 Monoclinic, P21/c
Hall symbol: -P 2ybc
a = 5.8389 (12) Å
b = 7.6865 (15) Å
c = 14.124 (3) Å
β = 97.90 (3)°
V = 627.9 (2) Å3 Z = 2
F(000) = 260
Dx = 1.303 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 5725 reflections
θ = 3.0–27.5°
µ = 0.39 mm−1 T = 293 K Needle, colorless 0.80 × 0.23 × 0.10 mm
Data collection
Rigaku R-AXIS RAPID IP area-detector diffractometer
Radiation source: rotating anode Graphite monochromator
ω scans
Absorption correction: multi-scan (ABSCOR; Higashi, 1995)
Tmin = 0.745, Tmax = 0.961
5725 measured reflections 1418 independent reflections 1180 reflections with I > 2σ(I)
Rint = 0.058
θmax = 27.5°, θmin = 3.0° h = −6→7
k = −9→9
l = −18→18
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.042 wR(F2) = 0.131 S = 1.03 1418 reflections 74 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0735P)2 + 0.1339P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.48 e Å−3
Δρmin = −0.28 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
S1 0.82869 (8) 0.07821 (8) 0.08636 (3) 0.0597 (3)
C1 0.6091 (3) −0.0518 (3) 0.01381 (14) 0.0574 (5)
H2A 0.6685 −0.0906 −0.0435 0.069*
H2B 0.5743 −0.1540 0.0495 0.069*
C2 0.7676 (3) 0.0563 (2) 0.20445 (12) 0.0426 (4)
C3 0.5774 (3) −0.0280 (3) 0.23236 (13) 0.0526 (5)
H6A 0.4668 −0.0775 0.1866 0.063*
C4 0.5528 (4) −0.0383 (3) 0.32789 (14) 0.0582 (5)
H4A 0.4259 −0.0961 0.3461 0.070*
C5 0.7124 (4) 0.0353 (3) 0.39639 (14) 0.0626 (5)
H5A 0.6942 0.0280 0.4606 0.075*
C6 0.8993 (4) 0.1200 (3) 0.36909 (15) 0.0683 (6)
H7A 1.0083 0.1702 0.4152 0.082*
C7 0.9276 (3) 0.1314 (3) 0.27418 (14) 0.0554 (5)
H3A 1.0548 0.1899 0.2567 0.066*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
S1 0.0474 (3) 0.0904 (5) 0.0429 (3) −0.0134 (2) 0.0117 (2) −0.0026 (2)
C1 0.0568 (11) 0.0718 (12) 0.0446 (10) 0.0078 (9) 0.0109 (8) −0.0094 (8)
C2 0.0400 (9) 0.0471 (8) 0.0407 (9) 0.0019 (7) 0.0053 (6) −0.0008 (6)
C3 0.0514 (10) 0.0626 (11) 0.0438 (9) −0.0124 (9) 0.0067 (7) −0.0045 (8)
C4 0.0639 (12) 0.0599 (10) 0.0535 (11) −0.0083 (10) 0.0181 (9) 0.0047 (8)
C5 0.0802 (14) 0.0698 (12) 0.0380 (9) 0.0040 (11) 0.0084 (9) 0.0014 (8)
C6 0.0719 (14) 0.0801 (14) 0.0484 (11) −0.0084 (11) −0.0076 (9) −0.0107 (10)
C7 0.0474 (10) 0.0631 (11) 0.0543 (11) −0.0094 (8) 0.0019 (8) −0.0032 (8)
supporting information
sup-3 Acta Cryst. (2005). E61, o2482–o2483
C2—C3 1.389 (2) C6—H7A 0.9300
C3—C4 1.379 (3) C7—H3A 0.9300
C2—S1—C1 105.15 (9) C5—C4—C3 120.99 (18)
C1i—C1—S1 111.06 (18) C5—C4—H4A 119.5
C1i—C1—H2A 109.4 C3—C4—H4A 119.5
S1—C1—H2A 109.4 C4—C5—C6 119.22 (18)
C1i—C1—H2B 109.4 C4—C5—H5A 120.4
S1—C1—H2B 109.4 C6—C5—H5A 120.4
H2A—C1—H2B 108.0 C5—C6—C7 120.75 (19)
C7—C2—C3 118.66 (17) C5—C6—H7A 119.6
C7—C2—S1 115.38 (14) C7—C6—H7A 119.6
C3—C2—S1 125.96 (14) C6—C7—C2 120.40 (18)
C4—C3—C2 119.97 (17) C6—C7—H3A 119.8
C4—C3—H6A 120.0 C2—C7—H3A 119.8
C2—C3—H6A 120.0
C2—S1—C1—C1i 85.4 (2) C3—C4—C5—C6 −0.2 (3)
C1—S1—C2—C7 173.98 (14) C4—C5—C6—C7 0.0 (3)
C1—S1—C2—C3 −5.93 (18) C5—C6—C7—C2 −0.4 (3)
C7—C2—C3—C4 −1.1 (3) C3—C2—C7—C6 0.9 (3)
S1—C2—C3—C4 178.82 (15) S1—C2—C7—C6 −178.97 (16)
C2—C3—C4—C5 0.7 (3)