Acta Cryst.(2002). E58, m721±m723 DOI: 10.1107/S1600536802020561 Svetlana N. Ivashevskajaet al. [Cu2Cl4(C9H7NO)2]
m721
metal-organic papers
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
Bis(
l
-quinoline
N
-oxide-
j
2O
:
O
)bis[dichloro-copper(II)], a powder diffraction study
Svetlana N. Ivashevskaja,a* Lyudmila A. Aleshina,a Vladimir P. Andreev,b Yakov P. Nizhnikband Vladimir V. Chernyshevc
aDepartment of Solid State Physics,
Petroza-vodsk State University, 185640 PetrozaPetroza-vodsk, Russia,bDepartment of Molecular Biology,
Biological and Organic Chemistry, Petrozavodsk State University, 185640 Petrozavodsk, Russia, andcChemistry Department, Moscow State
University, 119899 Moscow, Russia
Correspondence e-mail: [email protected]
Key indicators
Powder X-ray study
T= 293 K
Mean(C±C) = 0.04 AÊ
Rfactor = 0.064
wRfactor = 0.081
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2002 International Union of Crystallography Printed in Great Britain ± all rights reserved
The crystal structure of the title compound, [Cu2Cl4-(C9H7NO)2], has been determined from powder diffraction data. The crystals are built of dinuclear complex molecules, with a central four-membered Cu2O2 ring located about a crystallographic inversion centre, which involves two Cu atoms, each carrying two terminal chloro ligands each [CuÐ Cl = 2.213 (8) and 2.222 (8) AÊ] and two O atoms of the quinolineN-oxide ligand. The Cu atom has a square-planar coordination, with considerable distortions due to the
four-membered ring [OÐCuÐO = 78 (1) and ClÐCuÐCl =
100.2 (3)].
Comment
HeteroaromaticN-oxides are unique compounds, due to the fact that the NÐO group can act either as an electron-acceptor or an electron-donor, depending on the compound structure and conditions. Molecular complexes of hetero-aromaticN-oxides demonstrate a broad spectrum of biological activity (Ponomarenko, 1999). Moreover, adducts of quinoline
N-oxide with CuCl2 have interesting magnetic properties (Whymanet al., 1967).
The electronic spectrum of the title complex, (I), in ethanol is almost identical to the spectrum of quinoline N-oxide; however, in CH2Cl2, which is not capable of forming donor± acceptor bonds, a new band appears [379 (2.83) nm] and there is a decrease in band intensity. Such an effect is probably caused by symmetrization of the structure of the complex (decreasing log") and formation of new CuÐO bonds.
The intensities of the NÐO bands in the IR spectrum (1310 and 1272 cmÿ1) are decreased in (I), compared to the spec-trum of the parent quinolineN-oxide. On the other hand, in agreement with the literature ®ndings (Garveyet al., 1968), a new band at 1175 cmÿ1, caused by the formation of a donor± acceptor bond between the O atom of quinolineN-oxide and a Cu atom, appears; the 343±336 cmÿ1bands corresponding to the CuÐCl bonds are also present.
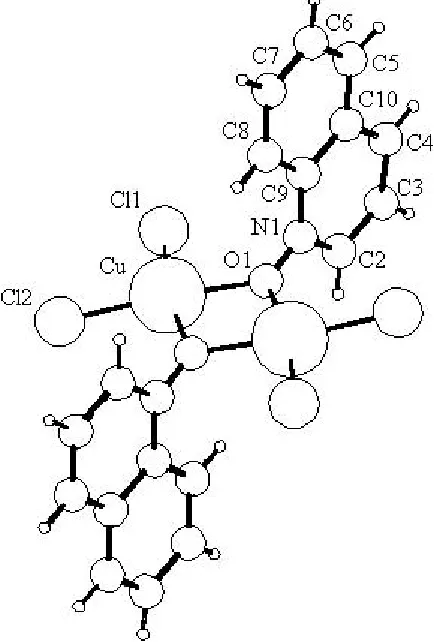
The structure of the dinuclear molecule consists of a central four-membered Cu2O2ring, formed by two Cu atoms and two
metal-organic papers
m722
Svetlana N. Ivashevskajaet al. [Cu2Cl4(C9H7NO)2] Acta Cryst.(2002). E58, m721±m723 quinolineN-oxide O atoms (Fig. 1). The structure is similar tothat of di--(pyridine N-oxide)-bis[dichlorocopper(II)] reported by Sageret al. (1967). The Cu atom has a distorted square-planar coordination, formed by two terminal chloro ligands [Cu belonging to quinolineN-oxide ligands [CuÐO1 = 2.11 (2) AÊ and CuÐO1i2.00 (2) AÊ; symmetry code: (i) 1ÿx, 1ÿy, 1ÿz]. The distortions of the coordination are revealed in the O1ÐCuÐO1i and Cl1ÐCuÐCl2 bond angles [78 (1) and 100.2 (3), respectively], which deviate signi®cantly from 90, as well as in the displacement of the Cu atom from the plane through its four ligands [0.21 (3) AÊ]. The plane of the Cu2O2ring forms a dihedral angle of 90 (2)with the quinoline fragment.
Experimental
Compound (I) was prepared in polycrystalline form, by mixing saturated ethanol solutions of quinolineN-oxide dihydrate (0.181 g, 1 mmol) and CuCl22H2O (0.171 g, 1 mmol). A yellow precipitate, which quickly turned black, was washed with ethanol and diethyl ether. It was then dried in air (yield: 58%). The electronic spectra of (I) in ethanol and in CH2Cl2were recorded using a Specord UV±Vis spectrometer. The IR spectra were measured in KBr using a Specord M-80 spectrometer.
Crystal data
[Cu2Cl4(C9H7NO)2]
Mr= 559.02
Monoclinic,P21=n
a= 11.780 (3) AÊ b= 14.872 (5) AÊ c= 6.061 (2) AÊ
= 98.27 (2) V= 1050.8 (6) AÊ3
Z= 2
Dx= 1.767 Mg mÿ3
FeKradiation
= 1.93728 AÊ
Cell parameters from 66 re¯ections
= 4.8±26.3
= 7.32 mmÿ1
T= 293 (2) K Black
Specimen shape: ¯at sheet 12122 mm
Particle morphology: no speci®c habit
Data collection
Burevestnic (Saint Petersburg) X-ray powder diffraction system DRON-4.07
Specimen mounting: the powder was sprinkled on the sample holder.
Specimen mounted in re¯ection mode
max= 35.0
h= 0!6 k= 0!8 l=ÿ3!3
2min= 10.00, 2max= 70.00 Increment in 2= 0.1
Re®nement
Re®nement onInet
Rp= 0.064
Rwp= 0.081
Rexp= 0.029
S= 2.80
Pro®le function: split-type pseudo± Voigt (Toraya, 1986)
77 parameters
H-atom parameters constrained
w= 1/Ymeas2 (/)max= 0.02
max= 0.52 e AÊÿ3
min=ÿ0.48 e AÊÿ3
Preferred orientation correction: spherical harmonics expansion (Ahteeet al., 1989) up to the 6th order
Table 1
Selected geometric parameters (AÊ,).
CuÐO1 2.11 (2)
CuÐO1i 2.00 (2) CuÐCl2CuÐCl1 2.213 (8)2.222 (8)
O1iÐCuÐO1 78 (1)
O1ÐCuÐCl2 168.8 (5)
O1ÐCuÐCl1 91.1 (5)
Cl2ÐCuÐCl1 100.2 (3)
CuiÐO1ÐCu 101.8 (1)
N1ÐO1ÐCu 126.3 (15)
Cl1ÐCuÐO1ÐN1 20 (2) CuÐO1ÐN1ÐC2 ÿ100 (2)
Symmetry code: (i) 1ÿx;1ÿy;1ÿz.
X-ray powder diffraction patterns were obtained with two X-ray powder instruments,viz.a Guinier±de Wolff camera and a DRON-4.07 diffraction system equipped with a standard resolution gonio-meter GUR-9 and scintillation counter. The ®rst pattern was used for indexing and the second pattern was measured for structure solution and re®nement. The powder was sprinkled on the sample holder to avoid preferred orientation. During the exposures, the specimen was spun in its plane to improve particle statistics. The unit-cell dimen-sions were determined using the indexing programTREOR(Werner Figure 2
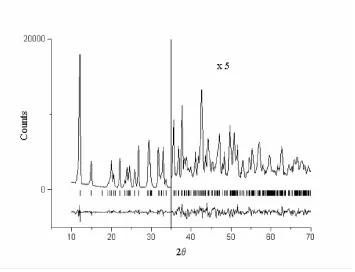
The Rietveld plot for (I), showing the observed and difference pro®les. The re¯ection positions are shown above the difference pro®le. Figure 1
et al., 1985) withM20= 39 andF29= 59 (0.0076, 65), using the positions of the ®rst 66 peaks.
The correct solution was found in monoclinic space groupP21/n. The structure was determined using theMRIAprogram (Zlokazov & Chernyshev, 1992), using grid search (Chernyshev & Schenk, 1998) and simulated annealing (Zhukov et al., 2001) techniques. The strength of the restraints was a function of the interatomic separation and, for intramolecular bond lengths, corresponds to an r.m.s. deviation of 0.03 AÊ. An additional restraint was applied to the planarity of the quinoline N-oxide fragment. Isotropic atomic displacement parameters were re®ned for Cu and Cl, and an overall
Uiso parameter was used for the remaining non-H atoms. H atoms were placed in geometrically calculated positions and allowed to re®ne using bond restraints, with a common isotropic displacement parameter Uiso(H) ®xed at 0.051 AÊ2. The diffraction pro®les are shown in Fig. 2.
Data collection: local program; cell re®nement:LSPAID(Visser, 1986); data reduction: local program; program(s) used to solve structure:MRIA(Zlokazov & Chernyshev, 1992); program(s) used to re®ne structure:MRIA; molecular graphics:PLATON(Spek, 2000); software used to prepare material for publication: MRIA,
SHELXL97 (Sheldrick, 1997) andPARST(Nardelli, 1983).
This work has been supported by the Russian Federation Educational Program `Integration' (project No. 1.5-100).
References
Ahtee, M., Nurmela, M., Suortti, P. & Jarvinen, M. (1989).J. Appl. Cryst.22, 261±268.
Chernyshev, V. V. & Schenk, H. (1998).Z. Kristallogr.213, 1±3.
Garvey, R. G., Nelson, J. H. & Ragsdale, R. O. (1968).Coord. Chem. Rev.N3, 375±407.
Nardelli, M. (1983).Comput. Chem.7, 95±98.
Ponomarenko, S. P. (1999). Plant Growth Regulators based on Pyridine Derivatives. Kiev: Technika. (In Russian.)
Sager, R. S., Williams, R. J. & Watson, W. H. (1967).Inorg. Chem.6, 951±955. Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Spek, A. L. (2000). PLATON for Windows. University of Utrecht, The
Netherlands.
Toraya, H. (1986).J. Appl. Cryst.19, 440±447. Visser, J. W. (1986).Powder Diffraction,1, 66±76.
Werner, P.-E., Eriksson, L. & Westdahl, M. (1985).J. Appl. Cryst.18, 367±370. Whyman, R., Copley, D. B. & Hat®eld, W. E. (1967).J. Am. Chem. Soc.89,
3135±3541.
Zhukov, S. G., Chernyshev, V. V., Babaev, E. V., Sonneveld, E. J. & Schenk, H. (2001).Z. Kristallogr.216, 5±9.
Zlokazov, V. B. & Chernyshev, V. V. (1992).J. Appl. Cryst.25, 447±451.
Acta Cryst.(2002). E58, m721±m723 Svetlana N. Ivashevskajaet al. [Cu2Cl4(C9H7NO)2]
m723
supporting information
sup-1
Acta Cryst. (2002). E58, m721–m723
supporting information
Acta Cryst. (2002). E58, m721–m723 [doi:10.1107/S1600536802020561]
Bis(
µ
-quinoline
N
-oxide-
κ
2O
:
O
)bis[dichlorocopper(II)], a powder diffraction
study
Svetlana N. Ivashevskaja, Lyudmila A. Aleshina, Vladimir P. Andreev, Yakov P. Nizhnik and
Vladimir V. Chernyshev
S1. Comment
Heteroaromatic N-oxides are unique compounds due to the fact that the N—O group can act either as an electron-acceptor or an electron-donor, depending on compound structure and conditions. Molecular complexes of heteroaromatic oxides demonstrate a broad spectrum of biological activity (Ponomarenko, 1999). Moreover adducts of quinoline N-oxide with CuCl2 have interesting magnetic properties (Whyman et al., 1967).
The electronic spectrum of the title complex, (I), in ethanol is almost identical to the spectrum of quinoline N-oxide; however, in CH2Cl2, which is not capable of forming donor–acceptor bonds, a new band appears [379(2.83)] and bands
decrease their intensities. Such an effect is probably caused by symmetrization of the structure of the complex (decreasing logε) and formation of new Cu—O bonds.
The intensities of the N—O bands in the IR spectra (1310 and 1272 cm−1) are decreased in (I) in comparison with the
spectrum of the parent quinoline N-oxide. On the other hand, in agreement with the literature findings (Garvey et al., 1968), a new bands at 1175 cm−1, caused by the formation of a donor–acceptor bond between the O atom of quinoline
N-oxide and Cu atoms, appears; the 343–336 cm−1 bands corresponding to the Cu–Cl bonds are also present.
The structure of the dimeric molecule consists of a binuclear complex with a central four-membered Cu2O2 ring, formed
by two Cu atoms and two quinoline N-oxide O atoms (Fig. 1). The structure is similar to that of di-m-(pyridine oxide)-bis[dichlorocopper(II)] reported by Sager et al. (1967). The Cu atom has a distorted square-planar coordination formed by two terminal chloro ligands [Cu—Cl1 = 2.222 (8) Å and Cu—Cl2 = 2.213 (8) Å] and two bridging O atoms belonging to quinoline N-oxide ligands [Cu—O1 = 2.11 (2) Å and Cu—O1i 2.00 (2) Å; symmetry code: (i) 1 − x, 1 − y, 1 − z]. The
distortions of the coordination are manifested in the O1—Cu—O1i and Cl1—Cu—Cl2 bond angles [78 (1) and
100.2 (3)°, respectively], which deviate significantly from 90°, as well as in the displacement of the Cu atom from the plane through its four ligands [0.21 (3) Å]. The plane of the Cu2O2 ring forms a dihedral angle of 90 (2)° with the
quinoline fragment.
S2. Experimental
Compound (I) was prepared in polycrystalline form by mixing saturated ethanol solutions of quinoline N-oxide dihydrate (0.181 g, 1 mmol) and CuCl2·2H2O (0.171 g, 1 mmol). A yellow precipitate, which quickly turned black, was washed
with ethanol and diethyl ether. It was then dried in air (yield: 58°). The electronic spectra of (I) were recorded using a Specord UV-Vis spectrometer in ethanol and in CH2Cl2. The IR spectra were measured in KBr using a Specord M-80
supporting information
sup-2
Acta Cryst. (2002). E58, m721–m723
S3. Refinement
X-ray powder diffraction patterns were obtained with two X-ray powder instruments, i.e. a Guinier-de Wolff camera and a DRON-4.07 diffraction system equipped with a standard resolution goniometer GUR-9 and scintillation counter. The first pattern was used for indexing and the second pattern was measured for structure solution and refinement. The powder was sprinkled on the sample to avoid preferred orientation. During the exposures, the specimen was spun in its plane to improve particle statistics. The unit-cell dimensions were determined using the indexing program TREOR (Werner et al., 1985) with M20 = 39 and F29 = 59 (0.0076, 65) using the positions of the first 66 peaks.
The correct solution was found in monoclinic space group P21/n. The structure was determined using the MRIA
program (Zlokazov & Chernyshev, 1992), using grid search (Chernyshev & Schenk, 1998) and simulated annealing (Zhukov et al., 2001) techniques. The strength of the restraints was a function of interatomic separation and, for intramolecular bond lengths, corresponds to an r.m.s. deviation of 0.03 Å. An additional restraint was applied to the planarity of the quinoline N-oxide fragment. Isotropic atomic displacement parameters were refined for Cu and Cl, and an overall Uiso parameter was used for the rest of non-H atoms. H atoms were placed in geometrically calculated positions
and allowed to refine using bond restraints, with a common isotropic displacement parameter Uiso(H) fixed at 0.051 Å2.
[image:5.610.197.414.312.633.2]The diffraction profiles are shown in Fig. 2.
Figure 1
supporting information
sup-3
[image:6.610.130.485.74.346.2]Acta Cryst. (2002). E58, m721–m723
Figure 2
The Rietveld plot for (I), showing the observed and difference profiles. The reflection positions are shown above the difference profile.
(I)
Crystal data
[Cu2Cl4(C9H7NO)2]
Mr = 559.02 Monoclinic, P21/n
a = 11.780 (3) Å
b = 14.872 (5) Å
c = 6.061 (2) Å
β = 98.27 (2)°
V = 1050.8 (6) Å3
Z = 2
F(000) = 556
Dx = 1.767 Mg m−3
Melting point: 208(1) K Fe Kα radiation, λ = 1.93728 Å
T = 293 K
Particle morphology: no specific habit black
flat_sheet, 12 × 12 mm
Data collection
X-ray powder diffraction system DRON-4.07 diffractometer
Radiation source: BSV-28, line-focus sealed tube
Pyrolitic graphite crystal monochromator
Specimen mounting: The powder was sprinkled on the sample holder.
Data collection mode: reflection Scan method: step
2θmin = 10°, 2θmax = 70°, 2θstep = 0.1°
Refinement
Refinement on Inet
Least-squares matrix: full with fixed elements per cycle
Rp = 0.064
Rwp = 0.081
Rexp = 0.029
χ2 = 7.840
601 data points
Profile function: split-type pseudo-Voigt (Toraya, 1986)
supporting information
sup-4
Acta Cryst. (2002). E58, m721–m723
H-atom parameters constrained
w = 1/Ymeas2
(Δ/σ)max = 0.02
Background function: Chebyshev polynomial up to the 5th order
Preferred orientation correction: Spherical harmonics expansion (Ahtee et al., 1989) up to the 6th order
Special details
Experimental. specimen was rotated in its plane
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Cu 0.6088 (3) 0.4852 (3) 0.3751 (5) 0.048 (1)* Cl1 0.6301 (5) 0.3910 (4) 0.0995 (12) 0.074 (3)* Cl2 0.7791 (6) 0.5509 (5) 0.4219 (9) 0.081 (3)* O1 0.4408 (15) 0.4384 (10) 0.384 (3) 0.087 (3)* N1 0.3980 (18) 0.3627 (13) 0.304 (4) 0.087 (3)* C2 0.330 (2) 0.3629 (18) 0.098 (5) 0.087 (3)* C3 0.286 (2) 0.283 (2) −0.002 (4) 0.087 (3)* C4 0.312 (2) 0.2013 (19) 0.107 (5) 0.087 (3)* C5 0.404 (2) 0.1175 (16) 0.427 (4) 0.087 (3)* C6 0.471 (2) 0.1138 (17) 0.636 (5) 0.087 (3)* C7 0.517 (2) 0.194 (2) 0.735 (4) 0.087 (3)* C8 0.492 (2) 0.2761 (16) 0.627 (4) 0.087 (3)* C9 0.424 (2) 0.2820 (18) 0.416 (4) 0.087 (3)* C10 0.380 (2) 0.2001 (19) 0.317 (4) 0.087 (3)*
H2 0.3132 0.4172 0.0207 0.051*
H3 0.2413 0.2844 −0.1423 0.051*
H4 0.2811 0.1476 0.0447 0.051*
H5 0.3740 0.0646 0.3580 0.051*
H6 0.4883 0.0587 0.7066 0.051*
H7 0.5617 0.1921 0.8752 0.051*
H8 0.5236 0.3290 0.6935 0.051*
Geometric parameters (Å, º)
Cu—O1 2.11 (2) C7—C8 1.40 (4)
Cu—O1i 2.00 (2) C9—N1 1.39 (3)
Cu—Cl2 2.213 (8) C9—C8 1.41 (3)
Cu—Cl1 2.222 (8) C9—C10 1.42 (4)
O1—N1 1.30 (2) C2—H2 0.94
C2—N1 1.38 (3) C3—H3 0.93
C2—C3 1.40 (4) C4—H4 0.93
C4—C3 1.40 (4) C5—H5 0.93
C4—C10 1.40 (4) C6—H6 0.93
C5—C6 1.40 (4) C7—H7 0.93
C5—C10 1.41 (4) C8—H8 0.94
supporting information
sup-5
Acta Cryst. (2002). E58, m721–m723
O1i—Cu—O1 78 (1) C5—C10—C9 121 (2)
O1i—Cu—Cl1 169.2 (6) C6—C5—C10 121 (2)
O1—Cu—Cl2 168.8 (5) C6—C7—C8 120 (3) O1i—Cu—Cl2 90.6 (5) C7—C8—C9 122 (2)
O1—Cu—Cl1 91.1 (5) C8—C9—C10 117 (2) Cl2—Cu—Cl1 100.2 (3) N1—C2—H2 120 Cui—O1—Cu 101.8 (1) C3—C2—H2 118
N1—O1—Cu 126.3 (15) C4—C3—H3 121
N1i—O1—O1 164.8 (5) C2—C3—H3 120
N1—C9—C8 124 (2) C3—C4—H4 120
N1—C9—C10 119 (2) C10—C4—H4 120
N1—C2—C3 121 (2) C6—C5—H5 120
O1—N1—C2 119 (2) C10—C5—H5 119
O1—N1—C9 121 (2) C5—C6—H6 120
C2—N1—C9 120 (2) C7—C6—H6 120
C3—C4—C10 120 (2) C6—C7—H7 120
C4—C3—C2 119 (3) C8—C7—H7 120
C4—C10—C5 119 (2) C7—C8—H8 120
C4—C10—C9 120 (2) C9—C8—H8 118
C5—C6—C7 119 (2)
Cl1—Cu—O1—N1 20 (2) Cu—O1—N1—C2 −100 (2)