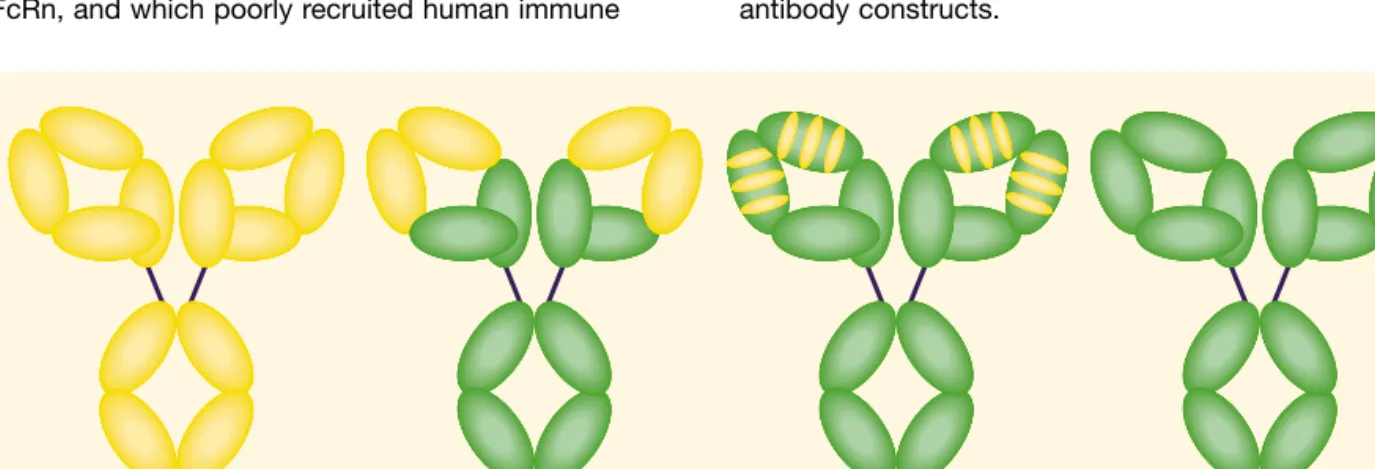
D
espite extensive efforts to achieve early diagno-sis and develop novel therapies, cancer has remained the second most common cause of death in developed countries.1More than a century ago, Paul Ehrlich speculated that the documented therapeutic efficacy of immune sera against infectious diseases may also be exploited to treat human malignancies.2This concept became feasible when Köhler and Milstein developed the technology to immortalize antibody-producing B cells, which allowed large-scale production of well-defined antibody reagents.3However, enthusiasm for this approach was disappointed by data from clinical trials in the early 1980s. Looking back at these studies today, their negative results were easily explained by the amounts of antibody, which were often too low to achieve tumor saturation. Furthermore, these trials were performed with murine antibodies that had short plasma half-lives, because of their insufficient interaction with human FcRn, and which poorly recruited human immuneeffector mechanisms and were also often immuno-genic. Over the following years, progress in molecular biology and protein chemistry allowed the generation of mouse/human chimeric antibodies initially, and later “humanized” antibodies. Today, competing technologies are available to generate fully human anti-bodies, which do not contain any murine sequences (Figure 1). With these novel types of reagents, antibody therapy has become an integral part of modern tumor therapy.
Today, five unconjugated antibodies have been approved for tumor therapy (Table), and many more are in different stages of preclinical and clinical devel-opment.4In the first part of this review, we will briefly summarize the clinical data that led to the approval of these antibodies. In the second part, we will discuss how an improved understanding of the relevant mechanisms of action of monoclonal antibodies may guide the development of improved second-generation antibody constructs.
2
Monoclonal antibodies for the treatment of cancer:
Reality and
future directions
MICHAEL DECHANT AND THOMAS VALERIUS, DIVISION OF NEPHROLOGY,DEPARTMENT OF MEDICINE I, UNIVERSITY SCHLESWIG-HOLSTEIN, CAMPUS KIEL, KIEL, GERMANY
Murine antibody Mouse/human
chimeric antibody
Humanized antibody Human antibody
Figure 1. Schematic diagram of murine, mouse/human chimeric, humanized, and fully human antibodies. Murine and human sequences are displayed in yellow and green, respectively.
was hampered by severe side effects in early clinical trials. Subsequently, alemtuzumab was investigated in the treatment of chronic lymphocytic leukemia (CLL), in which remarkable response rates were observed in chemotherapy-refractory patients.7These results led to FDA approval in 2001. Compared with rituximab, which also has activity in CLL if its dosing schedule is intensi-fied, application of alemtuzumab bears a significantly higher risk for opportunistic infections (especially cyto-megalovirus and herpes simplex virus). Alemtuzumab has also, however, activity against T-cell-derived malig-nancies.
Antibodies against solid tumors
Solid tumors are far more common than lymphomas, and therapeutic options for metastasized patients are often more limited. However, potential target antigens for antibody therapy were less well defined than on hematolymphatic malignancies. Among the best evalu-ated antigens were HER-2/neu and epidermal growth factor receptor (EGF-R), which are both members of the epidermal growth factor receptor family of tyrosine kinases.8Overexpression due to gene amplification or mutations of these two membrane antigens were shown to be involved in the malignant phenotype of many solid tumors and were correlated with a worse prognosis of patients, making both antigens attractive targets for immunotherapy.
Trastuzumab (Herceptin®), a humanized IgG1 antibody against HER-2/neu, was approved for the treatment of metastatic HER-2/neu-positive breast cancer patients. As only approximately 20–30% of ANTIBODIES APPROVED FOR CANCER THERAPY
Antibodies for lymphoma therapy
Lymphomas appeared to be particularly attractive targets for the development of therapeutic antibodies, because many antibodies against well-defined, lineage-specific antigens were already available, and
therapeutic antibodies were expected to reach their lymphoma targets more easily than they could in solid tumors. CD20 was considered as an almost ideal target antigen, because it is constitutively and homogenously expressed by the vast majority of B-cell lymphomas, but not on non-B cells, and CD20 is not shed nor internalized. Furthermore, antibodies against CD20 triggered apoptosis as well as complement-dependent and natural killer (NK) cell-dependent tumor cell killing. Consequently, a chimeric mouse/human immuno-globulin (Ig)G1 CD20 antibody – rituximab (Rituxan/ Mabthera®) – was the first cancer antibody to be approved by the US Federal Drug Administration (FDA) in 1997. A 4-week course of rituximab achieved objective responses in approximately 50% of heavily pretreated indolent lymphoma patients.5Toxicity of this novel regimen was low compared with chemotherapy and, interestingly, diminished with subsequent antibody applications. In patients with aggressive lymphomas, rituximab was combined with standard CHOP
(cyclophosphamide, adriamycin, vincristine, prednisone) chemotherapy (R-CHOP). This R-CHOP combination prolonged progression-free and overall survival compared with CHOP alone6– an improvement that had not been achieved with more sophisticated and aggressive chemotherapy regimens before. Meanwhile, rituximab has become an integral part of the manage-ment of almost all B-cell lymphoma patients.
Alemtuzumab (Campath®) is a humanized antibody against the CD52 antigen, which is expressed by the majority of lymphocytes. Alemtuzumab was originally developed as an immunosuppressive reagent for patients with rheumatoid arthritis, but its development
3
Generic name Proprietary name Target antigen Isotype Indication Date of US FDA approval
Rituximab Rituxan/Mabthera CD20 Chimeric IgG1 B-cell lymphoma November 1997
Alemtuzumab Campath CD52 Humanized IgG1 B-CLL May 2001
Trastuzumab Herceptin Her2/neu Humanized IgG1 Breast cancer September 1998
Cetuximab Erbitux EGF-R Chimeric IgG1 Colorectal February 2004
Bevacizumab Avastin VEGF Humanized IgG1 Colorectal February 2004
FDA, Food and Drug Administration; Ig, immunoglobulin; CLL, chronic lymphocytic leukemia; EGF-R, epidermal growth factor receptor; VEGF, vascular endothelial growth factor.
Table. Approved monoclonal antibodies for cancer therapy.
Antibody therapy has become
an integral part of modern
tumor therapy
breast cancer overexpress HER-2/neu, documentation of overexpression by immunohistochemistry or fluores-cence in situ hybridization (FISH) is mandatory before trastuzumab therapy is initiated. In strongly HER-2/neu-positive patients, however, addition of trastuzumab to standard chemotherapy (either taxol or anthracycline/ cyclophosphamide) was demonstrated to prolong event-free and overall survival.9
Overexpression of EGF-R is found on many common solid tumors, such as colon, non-small-cell lung and breast cancer. In some of these cancers, specific mutations of EGF-R were demonstrated to trigger continuous kinase activation, which may be involved in the malignant phenotype. Cetuximab (Erbitux®) was the first FDA-approved EGF-R-directed antibody, because cetuximab demonstrated significant therapeutic activity in irinotecan-refractory metastatic colorectal cancer patients.10Other EGF-R-directed antibodies are in different stages of clinical development.
Inhibition of tumor angiogenesis has long been con-sidered a valid approach against solid and hematologic malignancies,11and many small molecule inhibitors of angiogenesis have entered clinical evaluation. However, it was the antiangiogenic antibody bevacizumab (Avastin®), directed against vascular endothelial growth factor (VEGF), which became the first FDA-approved antiangiogenic compound. Addition of bevacizumab to chemotherapy with irinotecan, fluorouracil, and leukovorin significantly prolonged survival of patients with metastatic colorectal cancer compared with patients treated with chemotherapy alone.12
NOVEL APPROACHES
Currently approved antibodies were selected for clinical studies based on limited preclinical evaluation. Over the last decade, more refined antibody generation and production technology, and more sophisticated screening procedures became available to identify novel targets and to optimize antibody constructs for clinical development. For example, pertuzumab is a second-generation HER-2/neu antibody, which binds to the dimerization motif of HER-2/neu and thereby prevents HER-2/HER-2 homo- and HER-2/EGF-R or HER-2/HER-3 heterodimerization.13HuMaxCD20 is a
4
Michael Dechant obtained his MD degree from the University of Erlangen-Nürnberg, Germany. Currently, he is working at the Department of Medicine I at the University of Schleswig-Holstein, Campus Kiel, Germany. His main focus of research is to evaluate the therapeutic potential of human immunoglobulin A antibodies. E-mail: [email protected]
Thomas Valeriusstudied medicine at the University of Erlangen-Nürnberg, Germany. After his training in internal medicine, he specialized in hematology/oncology and in rheumatology. Since May 2005, he has been working at the University of Schleswig-Holstein, Campus Kiel, Germany. His main scientific interest has been the effector function of monoclonal antibodies in tumor therapy. E-mail: [email protected]
More refined antibody generation
and production technology, and
more sophisticated screening
procedures became available
over the last decade
fully human CD20 antibody, which binds to a different CD20 epitope than rituximab, and which is significantly more potent in activating human complement.14 Although proof is currently lacking that these and other novel reagents will therapeutically out-perform antibod-ies approved today, clinical studantibod-ies have been started, and mature results are eagerly awaited.
Novel approaches to improved antibody constructs also derive from studies into the mechanism of action of currently approved antibodies. Plenty mechanisms of action have been described in vitro (Figure 2), but the in vivo relevance of most of these mechanisms is large-ly unknown. Conceptuallarge-ly, antibodies’ mechanisms of action can be divided into direct effects that are medi-ated by the variable region of antibodies, and indirect mechanisms that require interaction of antibodies’ constant regions with either complement or with cellular Fc receptors. For rituximab, which is the most widely explored antibody in this regard, increasing evidence points to the importance of Fcγreceptors for clinical efficacy. For example, rituximab lost most of its therapeutic activity in mice deficient in activating Fcγ receptors, while its efficacy was enhanced in mice lacking the inhibitory FcγRIIb isoform.15In patients, a genetic polymorphism in the human FCGRIIIAgene critically determined treatment outcome after rituximab
therapy. Thus, patients homozygous for the 158V allele of the FcγRIIIa receptor, which binds human IgG1 more strongly than the 158F allele, demonstrated significantly better response rates to rituximab compared with homozygous 158F donors.16
Engineering IgG antibodies
Most of the antibodies currently used are of the human IgG1 isotype, because IgG1 effectively triggers anti-body-dependent cell-mediated cytotoxicity (ADCC) via FcγRIIIa on NK cells, activates human complement, and has an extended plasma half-life via binding to human FcRn. However, human IgG1 also binds to Fcγ recep-tors on noncytotoxic cells (such as FcγRIIa on platelets and B cells), to Fcγreceptors that do not activate effector cells (such as FcγRIIIb on polymorphonuclear leukocytes [PMN]), and to Fcγreceptors that even inhibit cell activation (such as FcγRIIb on monocytes/ macrophages). High-resolution mapping of the inter-action sites of Fcγreceptors with the IgG1 Fc region led to the identification of individual amino acids, which were critical for the selective binding of IgG1 mutants to individual Fcγreceptors.17Therefore, several approaches aim to improve antibody efficacy by increasing binding affinity to select activating isoforms (such as FcγRIIIa) and reducing interaction with the
5
Blockade of ligands
ADCC
Tumor cell
Apoptosis Growth arrestEffector cell
Signaling
Complement
Antibody Complement Fc receptor Target antigen LigandFigure 2. Potential mechanisms of action of therapeutic antibodies for cancer therapy. The relative contribution of these different mechanisms to the in vivo efficacy of therapeutic antibodies in patients is controversial. ADCC, antibody-dependent cell-mediated cytotoxicity.
inhibitory isoform (FcγRIIb). Thus, IgG1 variants of herceptin with improved binding to FcγRIIIa significantly enhanced ADCC with monocytes and NK cells in vitro. Interestingly, ADCC enhancement by some of these mutants was more significant with effector cells from donors carrying the FcγRIIIa-158F allele than with FcγRIIIa-158V donors. Consequently, these mutants could, to some degree, lead to more equal therapeutic efficacy of monoclonal antibodies in the different FcγRIIIa patient populations. Furthermore, mutants with increased affinity to FcRn were identified, which are supposed to have extended plasma half-lives com-pared with unmutated human IgG1. However, the in vivo relevance of all these mutants has not yet been determined.
In addition to their protein structure, antibodies con-tain complex carbohydrates, which are linked to cercon-tain amino acids and which are heterogeneous, depending on the cell system in which the antibodies were pro-duced. In IgG antibodies, the carbohydrate N-linked to asparagine (Asn) at position 297 was shown to be critically involved in Fcγreceptor binding. Thus, IgG variants deglycosylated at this position completely lost their capacity to interact with Fcγreceptors.18This Asn297 carbohydrate comprises a core oligosaccharide with heterogeneously attached monosaccharides. During the last few years, intensive efforts have been made to engineer these oligosaccharides to optimize Fc-Fcγreceptor interactions. Two approaches emerged as the most promising: the Lec13 subline of Chinese ovarian hamster (CHO) cells, the most frequently used antibody production cell line, is deficient in its ability to add fucose, but the remaining glycosylation of anti-bodies is very similar to that in humans.19In another approach, a specific glycosyltransferase (GnTIII) is cotransfected into the antibody-producing cell line, which adds a bisecting sugar to the core oligosaccha-ride and thereby prevents fucosylation of therapeutic IgG.20Interestingly, binding of fucose-deficient mutants to NK cells via FcγRIIIa was significantly increased compared with regularly fucosylated antibodies, and consequently, their cytotoxic potential was strongly enhanced. Again, clinical data with glyco-engineered antibodies are eagerly expected.
Bispecific antibodies
Due to the high homology and redundancy in the human Fcγreceptor system,21engineering of antibody mutants with increased affinity for individual Fcγ recep-tor isoforms proved norecep-toriously difficult. Bispecific antibodies constitute another approach. They typically consist of two antibody fragments; one specific for a target structure on tumor cells and a second directed against an activating receptor on effector cells. Thus, depending on the desired effector cell population,
specific trigger molecules can be engaged; for example FcγRIII (CD16) on NK cells, FcγRI (CD64) or FcαRI (CD89) on PMN and monocytes/macrophages, or CD3 on preactivated T cells. First-generation bispecific anti-bodies were produced as hybrid-hybridoma antianti-bodies, or by chemically cross-linking two F(ab’) fragments. In vitro and in vivo studies with these molecules confirmed their therapeutic potential and provided proof-of-principle for the concept. However, in phase I/II clinical studies, their limitations also became apparent;22both formats required expensive production and purification procedures, limiting optimal dosing. Furthermore, murine components in the constructs were immuno-genic. Additionally, hybrid-hybridoma antibodies induced Fc-mediated toxicity, while the plasma half-lives of chemically linked bispecific antibodies were only in the range of several hours. In the future, some of these problems may be solved by advances in protein engineering and expression technologies.23
IgA antibodies
Human IgG1 antibodies do not effectively activate PMN, which are the most numerous cytotoxic effector cell population in humans. Comparative studies with bispecific antibodies directed against FcαRI (CD89), FcγRI (CD64), or FcγRIII (CD16) demonstrated that optimal PMN recruitment was obtained with Fcα R-directed constructs.24FcαRI (CD89) is the myeloid receptor for IgA antibodies, which is constitutively expressed on PMN, monocytes/macrophages, and some types of dendritic cells, but – importantly for tumor immunotherapy – not on noncytotoxic cells. The activation of FcαRI by its natural ligands was investigated using panels of class-switched human leukocyte antigen (HLA) class II- or EGF-R- directed chimeric antibodies. In these studies, improved PMN recruitment was observed with IgA1 or IgA2 constructs compared with their IgG counterparts.25Furthermore, in vivo stimulation of donors with the myeloid growth factors granulocyte colony-stimulating factor or granulocyte-macrophage colony-stimulating factor significantly enhanced ADCC via IgA antibodies. Although IgA is the most abundantly produced anti-body isotype in humans and serves important functions in mucosal immunity,26its potential for immunotherapy has not been explored.27
6
Molecularly engineered
antibody constructs promise
further improvements in
CONCLUSIONS
Today, monoclonal antibodies constitute a novel approach in tumor therapy. The definite role of most currently approved antibodies in the routine manage-ment of cancer patients needs to be further defined in well-designed clinical trials. Antibodies against numer-ous innovative target antigens are in different stages of development, while second-generation antibodies against established targets have already entered the clinical arena. At the same time, molecularly engineered antibody constructs promise further improvements in antibody efficacy. Thus, significant therapeutic advances may well be expected over the next few years.
REFERENCES
1Bailar III JC, Gornik HL. Cancer undefeated. N Engl J Med 1997; 336: 1569–74.
2Ehrlich P. Beiträge zur Kenntnis der granulierten Bindegewebszellen und der eosinophilen Leukozyten. Arch Anat Physiol (Physiol Abt) 166, 1897
3Köhler G, Milstein C. Continuous culture of fixed cells secreting antibody of predefined specificity. Nature 1975; 256: 495–7. 4Glennie MJ, van de Winkel JGJ. Renaissance of cancer therapeutic
antibodies. Drug Discovery Today 2003; 8: 503–10.
5McLaughlin P, Grillo-Lopez AJ, Link BK, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to four-dose treatment program. J Clin Oncol 1998; 16: 2825–33.
6Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large B-cell lymphoma. N Engl J Med 2002; 346: 235–42. 7Rai KR, Freter CE, Mercier RJ, et al. Alemtuzumab in previously
treated chronic lymphocytic leukemia patients who also had received fludarabin. J Clin Oncol 2002; 20: 3891–7.
8Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990; 61: 203–12.
9Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpress HER2. N Engl J Med 2001; 344: 783–92. 10Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy
and cetuximabplus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004; 351: 337–45.
11Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971; 285: 1182–6.
12Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leukovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–42.
13Agus DB, Gordon MS, Taylor C, et al. Phase I clinical study of pertuzumab, a novel HER dimerization inhibitor, in patients with advanced cancer. J Clin Oncol 2005; 23: 2534–43.
14Teeling J, French RR, Cragg MS, et al. Characterisation of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin’s lymphoma. Blood 2004; 104: 1793–1800. 15Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc
receptors modulate in vivo cytotoxicity against tumor targets. Nat Med 2000; 6: 443–6.
16Cartron G, Dacheux L, Salles G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood 2002; 99: 754–8.
17Shields RL, Namenuk AK, Hong K, et al. High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J Biol Chem 2000; 276: 6591–6604.
18Jefferis R, Lund J, Pound JD. IgG-Fc-mediated effector functions: molecular definition of interaction sites for effector ligands and the role of glycosylation. Immunol Rev 1998; 163: 59–76.
19Shields RL, Lai J, Keck R, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular cytotoxicity. J Biol Chem 2002; 277: 26733–40.
20Umana P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol 1999; 17: 176–80.
21Van de Winkel JGJ, Anderson CL. Biology of human
immunoglobulin G Fc receptors. J Leukoc Biol 1991; 49: 511–24. 22Van Ojik HH, Valerius T. Preclinical and clinical data with bispecific
antibodies recruiting myeloid effector cells for tumor therapy. Crit Rev Oncol Hematol 2001; 38: 47–61.
23Peipp M, Valerius T. Bispecific antibodies targeting cancer cells. Biochem Soc Trans 2002; 30: 507–11.
24Valerius T, Stockmeyer B, van Spriel AB, et al. FcαRI (CD89) as a novel trigger molecule for bispecific antibodies. Blood 1997; 90: 4485–92.
25Dechant M, Vidarsson G, Stockmeyer B, et al. Chimeric IgA antibodies against HLA class II effectively trigger lymphoma cell killing. Blood 2002; 100: 4574–80.
26Mestecky J, Russell MW, Elson CO. Intestinal IgA: novel views on its function in the defence of the largest mucosal surface. Gut 1999; 44: 2–5.
27Dechant M, Valerius T. IgA antibodies for cancer therapy. Crit Rev Oncol Hematol 2001; 39: 69–77.
7
KEY MESSAGES
• Monoclonal antibodies are increasing the therapeutic options for tumor patients. • Antibody-dependent cell-mediated cytotoxicity
(ADCC) is considered a likely mechanism of action for many therapeutic antibodies.
• Approaches to enhance ADCC in vivo may further increase antibody efficacy.
• Antibodies against novel target antigens and ‘second-generation’ antibodies against established targets are expected to be approved over the next few years.
INTERESTING WEBSITES
Further information about topics discussed in this review can be found on the following websites:
Information regarding the latest approvals of monoclonal antibodies:
http://www.fda.gov/
Information concerning progress in antibody engineering: http://www.path.cam.ac.uk/~mrc7/mikeimages.html http://rzv054.rz.tu-bs.de/Biotech/SD/SDscFVSite.html http://www.biochem.unizh.ch/plueckthun/