Notch1 signaling in NOTCH1-mutated mantle cell lymphoma depends on Delta-Like ligand 4 and is a potential target for specific antibody therapy
Full text
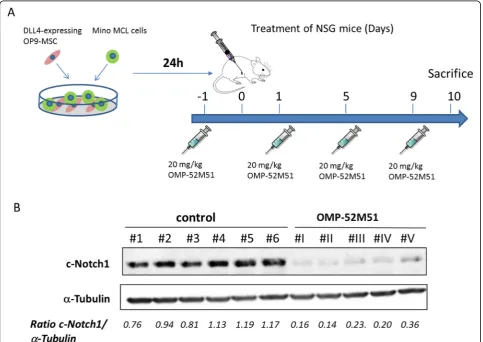
Figure
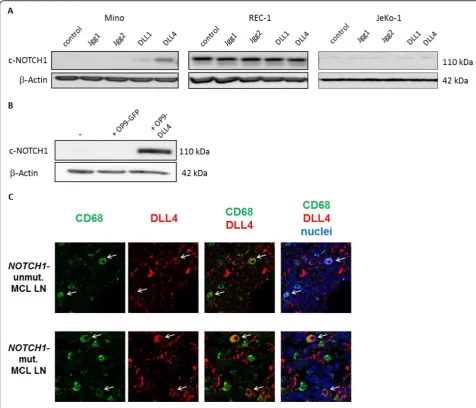
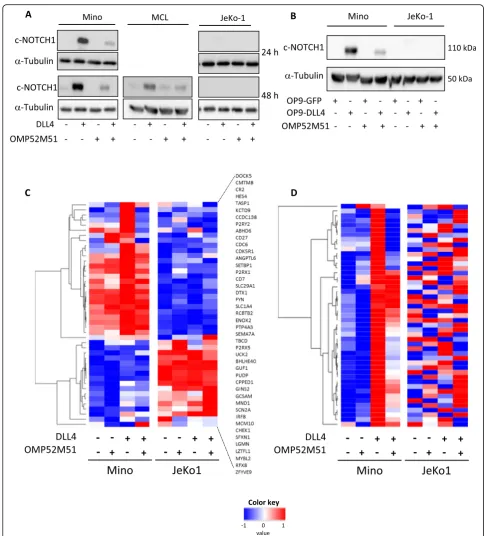
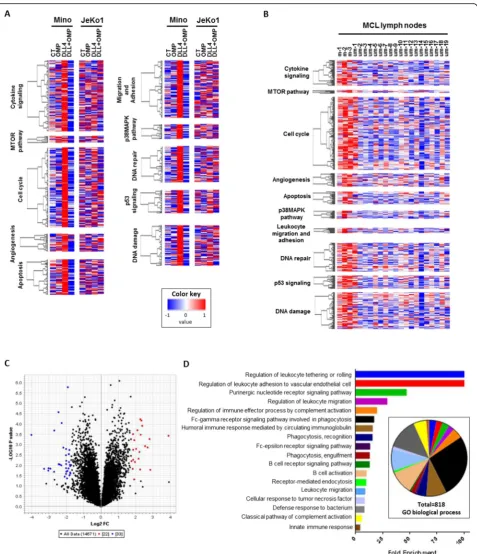
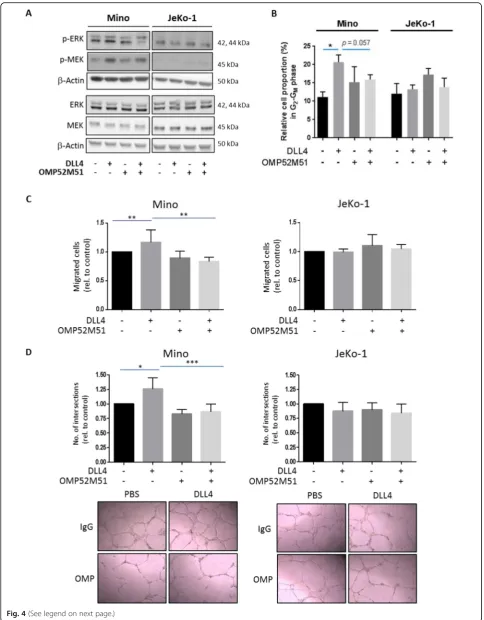
Related documents
International Journal of Scientific Research in Computer Science, Engineering and Information Technology CSEIT183715 | Received 05 Sep 2018| Accepted 18 Sep 2018 | September October 2018
TGF- β induced TMEPAI attenuates the response of mesenchymal type TNBC to doxorubicin and paclitaxel, but a minimal effect on cisplatin and bicalutamide.. This is an initial fi nding
Reactive Powder Concrete (RPC) is ultra high strength cementitious material composed of very fine powder with a maximum particle size of approximately 800µm and
To check the proposed methods we used an independent validation set consisting of the synthetic mixture solutions of HCT and AMH in the above working concentration
This fact is also consistent with the average angle of direction of 127.3° more closed to the optimum angle, than on the oxygen which has an average angle of 135.6°.Concerning the
Development and validation of UV spectrophotometric method for the determination of pazopanib hydrochloride in bulk and tablet formulation.. PAZ showed maximum sensitivity,
Abstract: A study has been carried out on Ta and Nb recovery by liquid-liquid extraction process using 4-methylacetophenone (4-MAcPh) as organic phase.. The 4-MAcPh was
[2] showed how to achieve resistance to second-order DPA (using a table re-masking method). Recent work has discussed affine masking [3], and a hardware-oriented masking scheme
