Abstract
SMITH, ANNE VOEGLER. Simulations of Protein Refolding and Aggregation Using
a Novel Intermediate-Resolution Protein Model. (Under the direction of Carol K. Hall.)
The objective of this thesis is to study the phenomena of amorphous and
or-dered protein aggregation. For this work, we developed an intermediate-resolution
protein model for use with the discontinuous molecular dynamics algorithm. With
this model, we simulated multi-protein systems at a greater level of detail than has
previously been possible and probed the energetic and structural characteristics of
amorphous and fibrillar protein aggregates.
We first developed an intermediate-resolution protein model and tested its
abil-ity to produce realistic protein dynamics. Each model residue consists of a
three-bead backbone and a single-three-bead side chain. Excluded volume, hydrogen bonds,
and hydrophobic interactions are represented by discontinuous potentials. Results
show that the model’s backbone motion is limited to realistic regions of φ-ψ con-formational space. In a series of simulations on different homopeptides, trends in
helicity as a function of residue type are found to be consistent with results from
previous studies. In simulations on a four-peptide system designed to produce a
four helix bundle, the resulting native structure is consistent with experimental and
previous simulation studies.
We then studied the competition between model protein refolding and
amor-phous aggregation for a model four helix bundle. Assembly of the bundle is found
to be optimal within a fixed temperature range, with the high-temperature boundary
a function of the complexity of the protein (or oligomer) to be folded and the
low-temperature boundary a function of the complexity of the protein’s environment.
As seen elsewhere, protein folding properties are strongly influenced by the
pres-ence of other proteins, and aggregates have substantial levels of native secondary
structure.
Simulations of Protein Refolding and
Aggregation Using a Novel
Intermediate-Resolution Protein Model
by
ANNE VOEGLER SMITH
A dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the Degree of Doctor of Philosophy
Chemical Engineering
Raleigh NC 27695-7905 2001
APPROVED BY:
Carol K. Hall Paul F. Agris
Chair of Advisory Committee
Robert M. Kelly Saad A. Khan
MacAdams. She received a B.Ch.E. degree in Chemical Engineering from Villanova
University in May, 1993 and a M.S. degree in Chemical Engineering from University of
Illinois at Urbana-Champaign in August, 1995. In August, 1995, she was admitted to
North Carolina State University to pursue graduate studies in Chemical Engineering.
iii
Acknowledgements
It is with pleasure that I acknowledge many of the people who have contributed
in various ways to the completion of this thesis.
Professor Carol K. Hall has been a dedicated and passionate guide in this
re-search endeavor.
Professors Ken Dill (University of California at San Francisco) and Yaoqi Zhou
(University of Buffalo) have been valuable sources of information and guidance over
the course of this research. I also thank Professors Sylvie Blondelle (Torrey Pines
Institute for Molecular Studies), Jeffery Kelly (Scripps Research Institute), and Dan
Kirschner (University of Pennsylvania) for their helpful discussions and advice on
our fibril aggregation studies.
The GAANN Biotechnology Fellowship of the U.S. Department of Education, the
National Institutes of Health, and the National Science Foundation have provided
generous financial support.
I thank the entire Hall research group, past and present, for a stimulating and
enjoyable work environment. Particular thanks go to the group system
administra-tors (Harpreet Gulati, Nirupama Kenkare, Julie McCormick, Brian Attwood, Andrew
Schultz) who have been saddled with the overwhelming responsibility of keeping
our equipment running and have consistently risen to the challenge.
Special thanks to Monica Hitchcock Lamm, Debbie Kaufman Follman, and Sean
Palecek for their wonderful friendship over the years.
I am especially grateful for the support of my family over the duration of this
thesis. My parents, Gerald and Georgia Voegler, have been a constant source of love
and support for which I will forever be thankful. I also thank my sister, Christine
MacAdams, and her husband, Mike, for their encouragement and for always offering
a humorous spin on what seemed, at times, to be a strange educational process.
Finally, I am grateful to my husband, Brad, whose love, encouragement, and patience
Page
List of Tables vii
List of Figures ix
Chapter 1 Introduction 1
1.1 References . . . . 7
Chapter 2 Bridging the Gap Between Homopolymer and Protein Models: A Discontinuous Molecular Dynamics Study 9 2.1 Introduction . . . . 9
2.2 Models and Methods . . . . 12
2.3 Results and Discussion . . . . 17
2.3.1 Chain Branching . . . . 17
2.3.2 Chain Heterogeneity . . . . 19
2.3.3 Chain Rigidity . . . . 21
2.3.4 Overlapping Beads . . . . 24
2.4 Conclusions . . . . 26
2.5 References . . . . 27
2.6 Figures . . . . 30
Chapter 3 α-Helix Formation: Discontinuous Molecular Dynamics on an Intermediate-Resolution Protein Model 50 3.1 Introduction . . . . 50
3.2 Models and Methods . . . . 57
3.2.1 Physical Chain Representation . . . . 57
v
3.2.3 Discontinuous Molecular Dynamics . . . . 63
3.2.4 Model Peptides. . . . 66
3.3 Results . . . . 66
3.3.1 Φ-Ψ Conformational Freedom . . . . 66
3.3.2 Polyalanine Helix Formation. . . . 67
3.3.3 Helix Formation as a Function of Side Chain Size . . . . 75
3.3.4 Polyglycine Dynamics. . . . 76
3.4 Discussion . . . . 77
3.5 Conclusions . . . . 78
3.6 References . . . . 80
3.7 Figures . . . . 88
Chapter 4 Assembly of a Tetrameric α-Helical Bundle: Computer Simula-tions on an Intermediate-Resolution Protein Model 109 4.1 Introduction . . . . 109
4.2 Models and Methods . . . . 114
4.2.1 Physical Chain Representation . . . . 114
4.2.2 Forces and Interactions . . . . 115
4.2.3 Discontinuous Molecular Dynamics . . . . 119
4.2.4 Model Peptides. . . . 121
4.3 Results and Discussion . . . . 122
4.3.1 Folding of an Amphipathicα-Helix . . . . 122
4.3.2 Assembly of a Tetramericα-Helical Bundle . . . . 127
4.4 Conclusions . . . . 135
4.5 References . . . . 137
4.6 Figures . . . . 141
Chapter 5 Protein Refolding Versus Aggregation: Computer Simulations on an Intermediate-Resolution Protein Model 154 5.1 Introduction . . . . 154
5.2.4 Model Peptides. . . . 168
5.3 Results and Discussion . . . . 169
5.4 Conclusions . . . . 178
5.5 References . . . . 180
5.6 Figures . . . . 185
Chapter 6 Simulation Studies of Small Model Fibrils 194 6.1 Introduction . . . . 194
6.2 Structure and Forces in the Model Fibril . . . . 196
6.3 Stability of the Model Fibril . . . . 200
6.4 Nucleation of the Model Fibril . . . . 203
6.5 Growth of the Model Fibril . . . . 206
6.6 Conclusions . . . . 208
6.7 References . . . . 209
6.8 Figures . . . . 212
Chapter 7 Future Work 220 7.1 Protein Model . . . . 220
7.2 Amorphous Aggregation . . . . 222
7.3 Ordered Aggregation . . . . 223
7.4 References . . . . 225
Appendices 226
vii
List of Tables
Page
Chapter 1 Introduction 1
Chapter 2 Bridging the Gap Between Homopolymer and Protein Models: A
Discontinuous Molecular Dynamics Study 9
2.1 Transition temperatures observed for each model studied. Superscript
adenotes rough estimates of temperature. . . . . 18
Chapter 3 α-Helix Formation: Discontinuous Molecular Dynamics on an
Intermediate-Resolution Protein Model 50
3.1 Simulation parameters: bead diameters, well diameters, bond lengths,
pseudobond lengths, and corresponding bond angles.. . . . 59 3.2 Helix properties. . . . . 73
Chapter 4 Assembly of a Tetrameric α-Helical Bundle: Computer
Simula-tions on an Intermediate-Resolution Protein Model 109
4.1 Simulation parameters: bead diameters, well diameters, bond lengths,
pseudobond lengths, and corresponding bond angles.. . . . 117 4.2 Occurrence of non-native andβhydrogen bonds during one- and
four-chain simulations within and below the optimal temperature range for
folding. . . . . 127 4.3 Native and non-native characteristics of misfolded structures in the
four-chain simulations. . . . . 134
5.2 Occurrence of non-native andβ hydrogen bonds during 1-, 4-, and 8-chain simulations within and below the optimal temperature range for
folding. . . . . 176 5.3 Native and non-native characteristics of aggregated structures. . . . . 177
Chapter 6 Simulation Studies of Small Model Fibrils 194
Chapter 7 Future Work 220
ix
List of Figures
Page
Chapter 1 Introduction 1
Chapter 2 Bridging the Gap Between Homopolymer and Protein Models: A
Discontinuous Molecular Dynamics Study 9
2.1 Model chain structures. . . . . 32 2.2 Bond lengths, bond angles, and bead sizes for models (d) through (g). 33
2.3 Reduced squared radius of gyration, reduced specific heat, and reduced
energy as a function of reduced temperature for the homopolymer. . 34 2.4 Reduced squared radius of gyration, reduced specific heat, and reduced
energy as a function of reduced temperature for the homoprotein. . . 35 2.5 A snapshot of the homopolymer chain at T∗=0.30. . . . . 36
2.6 A snapshot of the homoprotein chain at T∗=0.30. . . . . 37
2.7 A snapshot of the homoprotein chain at T∗=0.125. . . . . 38
2.8 Reduced squared radius of gyration, reduced specific heat, and reduced
energy as a function of reduced temperature for the heteroprotein. . 39 2.9 A snapshot of the heteroprotein chain at T∗=0.17. . . . . 40
2.10 Reduced squared radius of gyration, reduced specific heat, and reduced
energy as a function of reduced temperature for the homoprotein. . . 41 2.11 Non-uniform rigidity in models (d) through (g). . . . . 42 2.12 Snapshots of the rigid homoprotein chain atT∗ of 0.15 and atT∗ of
0.10. . . . . 43 2.13 Reduced squared radius of gyration, reduced specific heat, and reduced
energy as a function of reduced temperature for the rigid heteroprotein. 44
2.16 Snapshots of the overlapping-rigid homoprotein chain at T of 0.23 and atT∗of 0.10. . . . . 47
2.17 Reduced squared radius of gyration, reduced specific heat, and reduced
energy as a function of reduced temperature for the overlapping-rigid
heteroprotein. . . . . 48 2.18 Snapshots of the overlapping-rigid heteroprotein chain atT∗of 0.16. 49
Chapter 3 α-Helix Formation: Discontinuous Molecular Dynamics on an
Intermediate-Resolution Protein Model 50
3.1 An amino acid residue.. . . . 90 3.2 Covalent bonds. . . . . 91 3.3 Backbone hydrogen bonding. . . . . 92 3.4 Fluctuations in hydrogen bond NHO angle and O−H distance over
time.. . . . 93 3.5 Ramachandran plots for simulations of non-glycine residues and
glycine residues. . . . . 94 3.6 Percentage of α-helical hydrogen bonds formed for the 20-residue
polyalanine chain as a function of reduced temperature. . . . . 95 3.7 Number of α-helical hydrogen bonds formed versus reduced time for
a 20-residue polyalanine chain atT∗=0.10. . . . . 96
3.8 Snapshots of the polyalanine chain during the low-temperature run
shown in Figure 3.7. . . . . 97 3.9 Number of α-helical hydrogen bonds formed versus reduced time for
a 20-residue polyalanine chain atT∗=0.125. . . . . 98
3.10 Snapshots of the polyalanine chain during the high-temperature run
xi
3.11 Average number ofα-helical hydrogen bonds formed as a function of reduced time for a 20-bead polyalanine chain atT∗=0.077.. . . . 100
3.12 The “fast-folding” simulation of Figure 3.11 divided into 6 time blocks. 101
3.13 As in Figure 3.12, but for the “slow-folding” simulation of Figure 3.11. 102
3.14 The time that elapses beforeα-helical hydrogen bonds form. . . . . . 103 3.15 Average percentα-helical hydrogen bonds formed as a function of
re-duced temperature for polyalanine chains with 10, 20, and 30 4-bead
residues. . . . . 104 3.16 Average percentα-helical hydrogen bonds formed as a function of
re-duced temperature for chains with polyalanine-sized and larger side
chains. . . . . 105 3.17 Snapshots of the lowest-energy states of homogeneous peptide chains
with different size side chains. . . . . 106 3.18 Total number of hydrogen bonds formed and number ofα-helical
hy-drogen bonds formed for the polyglycine homopolymer over reduced
time.. . . . 107 3.19 Snapshots of the polyglycine homopolymer during the run shown in
Figure 3.18. . . . . 108
Chapter 4 Assembly of a Tetrameric α-Helical Bundle: Computer
Simula-tions on an Intermediate-Resolution Protein Model 109
4.1 An amino acid residue.. . . . 142 4.2 The 16-residue peptide in anα-helix. . . . . 143 4.3 Snapshots of the isolated 16-residue protein model during folding to
theα-helical native conformation. . . . . 144 4.4 Percentα-helical hydrogen bonds formed as a function of T* forHP=0,
HP=18HB,HP= 1
6HB,HP= 1
4HB, andHP= 1
2HB. . . . . 145
4.5 Snapshots of misfolded structures during simulations on isolated
residue chains atT =0.055.. . . . 148 4.8 Native structure of the tetramericα-helical bundle. . . . . 149 4.9 Nativeness parameter, Q, versus reduced temperature for the
four-chain simulations. . . . . 150 4.10 Snapshots of folding of a tetramericα-helical bundle. . . . . 151 4.11 Number of α-helical hydrogen bonds formed, number of inter-chain
hydrophobic contacts formed, and value of Q during the folding
tra-jectory of the tetramericα-helical bundle depicted in Figure 4.10. . . 152 4.12 Misfolded structures during simulations of four 16-residue chains at
T∗=0.072. . . . . 153
Chapter 5 Protein Refolding Versus Aggregation: Computer Simulations on
an Intermediate-Resolution Protein Model 154
5.1 An amino acid residue.. . . . 186 5.2 The 16-residue peptide in anα-helix. . . . . 187 5.3 Native structure of the tetramericα-helical bundle. . . . . 188 5.4 Nativeness parameter, Q, versus reduced temperature for the four- and
eight-chain simulations. . . . . 189 5.5 Snapshots of the conformations of each chain or complex of chains in
an eight-chain simulation that results in formation of two tetrameric
α-helical bundles. . . . . 190 5.6 Number of α-helical hydrogen bonds formed and number of
inter-chain hydrophobic contacts formed for the red-orange-yellow-magenta
xiii
5.7 Number ofα-helical hydrogen bonds formed and number of inter-chain hydrophobic contacts formed for the green-blue-purple-turquoise
tetramer during the folding trajectory of the two tetrameric α-helical bundles depicted in Figure 5.5. . . . . 192 5.8 An eight-chain aggregate in a simulation atT∗=0.076. . . . . 193
Chapter 6 Simulation Studies of Small Model Fibrils 194
6.1 Model fibril structure and interactions. . . . . 213 6.2 QHBand QTOT versus t* for the fibril and QHBfor the system ofα-helices. 214 6.3 Histogram of the normalized distribution of hydrogen bond lifetimes
for the model fibril complex and the system of α-helices during the simulations shown in Figure 6.2. . . . . 215 6.4 QHBand QHHversus t* for the reassembly of partially-unfoldedβ-sheet
complexes. . . . . 216 6.5 Number of fibrillar hydrogen bonds and hydrophobic contacts versus
t* during an unfolding simulation at T*=0.167 and during a refolding
simulation at T*=0.143. . . . . 217 6.6 (a) Plot of the highest values of QHH and QHB in a 4-residue 12-chain
block in each partially-unfolded configuration. (b) Partially-unfolded
configuration. . . . . 218 6.7 An amorphous aggregate. . . . . 219
Chapter 7 Future Work 220
Introduction
Protein aggregation is a serious problem in a wide range of industrial, research, and
medical settings.1–3In the biotechnology industry, intracellular aggregates called
in-clusion bodies often sequester the heterologous proteins of pharmaceutical interest,
requiring complicated de-aggregation procedures to recover active protein. Once the
active form of a protein-based drug has been recovered, drug packaging, shipping,
and storage can compromise its stability.4,5 In protein research, the tendency of
large, multi-protein structures to precipitate out of solution hinders investigations
of protein structure and function.1 In the medical field, many diseases including
Alzheimer’s,6Huntington’s,7 Creutzfeld-Jakob,8and cataracts9are characterized by
the presence of aberrant aggregates of normally soluble proteins. A thorough
un-derstanding of aggregate structure, the factors contributing to aggregate stability,
and the underlying mechanisms of protein aggregation is of immense importance
to the biotechnology, pharmaceutical, and biomedical industries.
Protein aggregates can be categorized by the presence or absence of order in
their structures; inclusion bodies are an example of disordered, or amorphous,
ag-gregates, and amyloid fibrils are an example of ordered aggregates.10 Although
in-clusion body formation is not fully understood, abnormally high protein
concentra-tions, errors in folding, and errors in protein-chaperone interactions are suspected
to contribute to formation.10Though little is known about the structure of inclusion
bodies or the processes by which they form, recent experimental11–13and simulation
studies14,15 suggest that they are composed of partially-native protein chains, not
2
long-range order and often display substantial beta-sheet structure.6In Alzheimer’s
disease victims, amyloid fibrils consisting of aggregated beta-amyloid proteins form
extracellular amyloid plaques that ravage the brain. Amyloid fibrils have been
ob-served in many different diseases and, though a different protein is associated with
each disease, exhibit strikingly similar morphologies.16,17 While high concentration
of protein and certain mutations have been identified as contributing to fibrillar
aggregate formation, the mechanisms of amyloid formation have not yet been
eluci-dated.10Despite rapid experimental advances and the clear necessity of
understand-ing aggregation and treatunderstand-ing its undesired consequences, fundamental questions
re-garding protein aggregation remain unanswered. In most cases, little is known about
the interactions responsible for stabilizing aggregates, the mechanisms responsible
for aggregation, or the effect of environmental conditions on aggregation.
Computer simulation is a powerful tool for studying the molecular level
struc-ture and interactions in systems of model proteins. Although simulations of
indi-vidual proteins are common, focusing largely on the protein folding process, very
few simulations of multi-protein systems have been performed. In addition, due
to limitations in computer power, all previous efforts to simulate protein
aggrega-tion in multi-protein systems have utilized very simplified, low-resoluaggrega-tion, lattice
protein models. One of the earliest computational approaches to study protein
aggregation involved two-dimensional Monte Carlo simulations on hexagons, with
the association of hexagons corresponding to protein aggregation.18,19 Our group
probed the importance of protein concentration and solvent quality on the
compe-tition between protein folding and amorphous aggregation using two-dimensional
Monte Carlo simulations on heteropolymer model proteins, where each protein is
represented by a flexible sequence of hydrophobic or polar spherical beads.14
Re-cently, a number of other groups have described exact enumeration studies and
Monte Carlo simulations on multi-protein systems modeled with the heteropolymer
chain model.15,20–22 Each of these studies offers insight into certain aspects of the
protein aggregation phenomenon, such as the structure of aggregating proteins or
protein model that enables detailed, yet computationally efficient, simulations of
multi-protein systems. Chapter 2 lays the foundation for the physical structure of
the protein model, Chapter 3 describes the creation of a suitable hydrogen bonding
potential for the model, and Chapter 4 describes the model hydrophobic potential.
In Chapter 5, we present results from simulations to study the aggregation of model
peptides into amorphous aggregates, analogous to inclusion bodies described above.
In Chapter 6, we present results from simulations to study the stability of ordered
aggregates, analogous to the fibril structures described above. A summary of each
of these chapters is given below. Finally, in Chapter 7, we suggest areas for further
study.
In Chapter 2, we build the foundation for a new, intermediate-resolution
pro-tein model from the widely-used simple, homopolymer chain model. A series of
seven off-lattice protein models is analyzed that spans a range of chain geometries
from the low-resolution homopolymer model to an intermediate-resolution model
that accounts for the presence of side chains, the varied character of the individual
amino acids, the rigid nature of protein backbone angles, and the length scales that
characterize real protein bead sizes and bond lengths. Discontinuous molecular
dynamics is used to study the transition temperatures and physical structures
asso-ciated with each protein model. Our results show that each protein model undergoes
multiple thermodynamic transitions that roughly correlate with protein transitions
during folding to the native state. Other realistic protein behavior, such as burial
of hydrophobic side chains and hindered motion due to backbone rigidity, is
ob-served with the more-detailed models. Despite their simplicity when compared with
all-atom protein models, the models presented in this chapter display a significant
amount of protein character.
In Chapter 3, we present the detailed structure of our intermediate-resolution
4
protein secondary structure formation. Physically, each model residue consists of
a detailed, three-bead backbone and a simplified, single-bead side chain. Excluded
volume and hydrogen bond interactions are constructed with discontinuous (i.e.
hard-sphere and square-well) potentials. Simulation results show that the model’s
backbone motion is limited to realistic regions ofφ-ψconformational space. Model polyalanine chains undergo a locally cooperative transition to formα-helices stabi-lized by backbone hydrogen bonding, while model polyglycine chains tend to adopt
non-helical structures. When side chain size is increased beyond a critical diameter,
steric interactions prevent formation of longα-helices. These trends in helicity as a function of residue type have been well documented by experimental, theoretical,
and simulation studies and demonstrate the ability of the intermediate-resolution
model developed in this work to accurately mimic real peptide behavior. The
effi-cient algorithm used allows observation of the complete helix-coil transition within
fifteen minutes on a single-processor workstation.
In Chapter 4, we add a hydrophobicity term to our intermediate-resolution
pro-tein model and perform discontinuous molecular dynamics simulations to study the
folding of an isolated, small model peptide to an amphipathicα-helix and the assem-bly of four of these model peptides into a four helix bundle. Simulations efficiently
sample conformational space allowing complete folding trajectories from random
initial configurations to be observed within 15 minutes for the one-peptide system
and within 15 hours for the four-peptide system on a 500MHz workstation. The
native structures of both theα-helix and the four helix bundle are consistent with experimental characterization studies and with results from previous simulations
on these model peptides. In both the one- and four-peptide systems, the native state
is achieved during simulations within an optimal temperature range, a phenomenon
also observed experimentally. The ease with which our simulations yield
reason-able estimates of folded structures demonstrates the power of the model developed
for this work and suggests that simulations of very long times and of multi-protein
systems are possible with this model.
folding trajectories from random initial configurations to two four-helix bundles
are possible within two days on a single processor workstation. Assembly of the
bundles follows two main pathways, one through a trimeric intermediate and the
other through an intermediate with two dimers. The proportion of trajectories that
follow each route is significantly different for the eight-peptide system than for the
four-peptide system described in Chapter 4, which suggests, as have our previous
simulations, that protein folding properties are strongly influenced by the presence
of other proteins. Folding of the bundles is optimal within a fixed temperature
range, with the high-temperature boundary a function of the complexity of the
pro-tein (or oligomer) to be folded and the low-temperature boundary a function of the
complexity of the protein’s environment. Above the optimal temperature range for
folding, the model chains tend to unfold; below the optimal range, the model chains
tend to aggregate. As has been seen previously, aggregates have substantial levels
of native secondary structure, suggesting that aggregates are composed largely of
partially-folded intermediates, not denatured chains.
In Chapter 6, we incorporate known physical characteristics of aggregate
struc-ture, which have emerged from experimental analyses in recent years, into a
hy-pothetical model fibril consisting of twelve 16-residue model polyalanine-based
peptides. Each model residue is made up of four beads, three representing the
residue backbone and one representing the residue side chain. Steric interactions,
hydrophobic interactions, and hydrogen bonding forces are explicitly represented
in the model. We perform discontinuous molecular dynamics simulations on this
model fibril to study its stability, nucleation, and growth. In our simulations, the
6
has been suggested by experimental studies, not of hydrogen bonds between
pep-tide backbones. We also find that hydrogen bonds within the model fibril are not as
stable as hydrogen bonds in modelα-helices. In fact, the average lifetime of a hy-drogen bond in the model fibril is less than 40% that in a dilute system ofα-helices. We detect a critical ordered substructure that must be present to ensure assembly
of the model fibril and determine the minimum number of model peptides required
for fibril nucleation. In fibrillar complexes with the proposed model, there is an
energetic preference forβ-sheet elongation as opposed toβ-sheet creation, provid-ing an explanation for why ordered protein aggregates in nature tend to grow very
asymmetrically.
Chapters 2 through 6 are adapted from the following publications:
Chapter 2: “Bridging the Gap Between Homopolymer and Protein Models: A
Discon-tinuous Molecular Dynamics Study”, Anne Voegler Smith and Carol K. Hall,Journal
of Chemical Physics, 2000, vol 113, pp. 9331-9342.
Chapter 3: “α-Helix Formation: Discontinuous Molecular Dynamics on an Intermediate- Resolution Protein Model”, Anne Voegler Smith and Carol K. Hall,
Pro-teins: Structure, Function, Genetics, accepted.
Chapter 4: “Assembly of a Tetramericα-Helical Bundle: Computer Simulations on an Intermediate- Resolution Protein Model”, Anne Voegler Smith and Carol K. Hall,
Proteins: Structure, Function, Genetics, accepted.
Chapter 5: “Protein Refolding Versus Aggregation: Computer Simulations on an
Intermediate-Resolution Protein Model”, Anne Voegler Smith and Carol K. Hall,
Jour-nal of Molecular Biology, submitted.
Chapter 6: “Simulation Studies of Small Model Fibrils”, Anne Voegler Smith and
[2] A. Mikraki and J. King, Biotechnol.,7, 690 (1989).
[3] J. King and S. Betts, Nat. Biotech., 17, 637 (1999).
[4] M. Manning, K. Patel, and R. Borchardt, Pharm. Res.,6, 903 (1989).
[5] H. R. Costantino, R. Langer, and A. M. Klibanov, Biotechnol.,13, 493 (1995).
[6] D. J. Selkoe, Nature,399, A23 (1999).
[7] F. Persichetti, Neurobiol. Dis.,6, 364 (1999).
[8] J. Tatzelt, R. Voellmy, and W. J. Welch, Cell Mol Neurobiol,18, 721 (1998).
[9] M. D. Perng, J. Biol. Chem.,274, 33235 (1999).
[10] A. L. Fink, Fold. Des.,3, R9 (1998).
[11] M. A. Speed, D. I. Wang, and J. King, Prot. Sci., 4, 900 (1995).
[12] R. Wetzel, Cell,86, 699 (1996).
[13] J. King, C. Haase-Pettingell, A. S. Robinson, M. Speed, and A. Mitraki, FASEB J.,
10, 57 (1996).
[14] P. Gupta, C. K. Hall, and A. C. Voegler, Prot. Sci.,7, 2642 (1998).
[15] R. A. Broglia, G. Tiana, S. Pasquali, H. E. Roman, and E. Vigezzi, Proc. Natl. Acad.
Sci. USA,95, 12930 (1998).
[16] M. Sunde, L. C. Serpell, M. Bartlam, P. E. Fraser, M. B. Pepys, and C. C. F. Blake,
J. Mol. Biol.,273, 729 (1997).
[17] R. Kisilevsky, J. Struct. Biol.130, 99 (2000).
8
[19] S. Y. Patro and T. M. Przybycien, Biophys. J.,70, 2888 (1996).
[20] S. Istrail, R. Schwartz, and J. King, J. Comput. Biol.,6, 143 (1999).
[21] G. Giugliarelli, C. Micheletti, J. R. Banavar, and A. Maritan, J. Chem. Phys.,113,
5072 (2000).
[22] P. M. Harrison, H. S. Chan, S. B. Prusiner, and F. E. Cohen, J. Mol. Biol.,286, 593
Bridging the Gap Between Homopolymer and
Protein Models: A Discontinuous Molecular
Dynamics Study
2.1 Introduction
We are engaged in a program of research aimed at investigating protein
aggre-gation, a phenomenon that causes serious problems in the biomedical and
phar-maceutical industries 1–5 and has been linked to a number of human conditions
including Alzheimer’s disease and cataracts.6,7 Despite its importance, protein
ag-gregation has received little attention from the theoretical community; and, as yet,
a general understanding of its physical basis is far from complete. To study
ag-gregation computationally, protein models must be developed that contain enough
genuine protein character to mimic real proteins yet are simple enough to allow
computer simulation of multi-protein systems over long times. In this paper, we
describe our efforts to construct a minimalist protein model that is computationally
tractable and could eventually serve as the basis for studies of protein aggregation.
The ability of idealized, or minimalist, models to provide insights into the
coarse-grained structure and folding mechanisms of proteins has long been
rec-ognized.8–13All-atom models are physically more realistic than minimalist models;
however, their complexity precludes their use for studies of long-time events.
Mini-malist models, on the other hand, have played an important role in theoretical and
10
the major physical changes experienced by a protein during folding from denatured
random conformations to the native state, albeit with a sacrifice in detail.14–20,23,24
Among the more popular minimalist models are the homo- and heteropolymer chain
models in which each amino acid residue is represented by a single sphere with
iden-tical (homo) or varied (hetero) interaction parameters.25–27 In addition to studies
of physical transitions during folding of the chain,20–22 homopolymer simulations
have been used to probe for molten globule intermediates during the globule-to-coil
transition28,29 and have successfully produced experimentally-characterized
struc-tures.30 Recently, heteropolymer models have gained popularity as they can more
accurately represent the character of real protein chains. Both lattice and
continu-ous models have been used to demonstrate that heteropolymers “fold” to a small
set of low energy (“native”) structures.8,14,15,31–35
Toward our goal of building a protein model suitable for simulations of protein
aggregation, we previously performed single-chain simulations of a freely-jointed,
tangent, square-well chain homopolymer model, a very simple protein model in
which each amino acid residue is represented by a sphere.21,22 Despite the
sim-plicity of the model, the system undergoes multiple equilibrium thermodynamic
transitions that roughly correlate with gas-to-liquid, liquid-to-solid, and
solid-to-solid transitions. Similar multiple phases are also observed during protein
fold-ing wherein proteins transition between denatured, collapsed globule, and native
states.36The gas-to-liquid collapse transition observed in homopolymer simulations
mimics the structural collapse in the early stages of protein folding that is often
driven by hydrophobic attraction. A homopolymer liquid-to-solid transition
qualita-tively corresponds to a protein transition from the collapsed, molten-globule state
to the native state. The third transition seen for homopolymers at very low
tem-peratures corresponds to complete solidification of the protein and may indicate a
cold-denaturation process to an inactive state.22That the simplest of chain models,
the homopolymer, can qualitatively describe the stages of protein folding prompted
us to consider the behavior of a range of simple protein models based on the
model. Our focus here is on developing a simple representation of protein
geome-try that accounts for the presence of bulky side chains, the varied character of the
individual amino acids, the rigid nature of protein backbone angles, and the length
scales that characterize real protein bead sizes and bond lengths. The
homopoly-mer model is modified in four steps by adding the following physical features piece
by piece: (A) branched structure, (B) heterogeneity, (C) realistic backbone angles,
and (D) realistic ratios of bead sizes to bond lengths. By building the model in a
step-by-step fashion, we create a hierarchy of distinct protein models that allows us
to assess the relative importance of these features as measured by their impact on
the phase transitions in the system. We perform discontinuous molecular
dynam-ics simulations on each of the protein models in this hierarchy at several different
temperatures, monitoring the radius of gyration, specific heat, internal energy, and
overall chain conformation. For each model studied, we report how the particular
modification affects the observed thermodynamic phase transitions and the
result-ing physical structures.
Highlights of our simulation results are the following. We find that introducing
branching to the homopolymer model has little effect on either the temperatures or
the strengths of the thermodynamic transitions. As with the homopolymer model,
branched homopolymers adopt symmetric conformations at moderately-low
tem-peratures and tight, spherical conformations at very lower temtem-peratures.
Hetero-geneity, however, causes both a downward temperature shift and a strengthening of
the individual transitions which can be attributed to a loss of constraints.
Heteroge-neous chains assume different types of compact conformations since only a fraction
of their segments are subject to hydrophobic collapse. In the systems that
incorpo-rate realistic backbone angles and realistic ratios of bead sizes to bond lengths, we
find multiple transitions similar to those obtained for the other models studied. The
12
chain rigidity. In a forthcoming paper, we define a potential energy function for
use with the most-realistic physical model developed here, and we show that this
physical model is indeed detailed enough to exhibit protein-like behavior but simple
enough to allow for simulations covering very long time scales.37
The remainder of the paper is organized as follows. Section 2.2 describes the
models studied and the discontinuous molecular dynamics simulation technique
used. In Section 2.3, we present the results associated with the four types of
modi-fications to the homopolymer model considered here: (A) chain branching, (B) chain
heterogeneity, (C) chain rigidity, and (D) bead overlap. Section 2.4 provides a brief
conclusion.
2.2 Models and Methods
We use discontinuous molecular dynamics (DMD) computer simulations to
study the effect of different chain geometries on the observed phase transitions
and types of equilibrium structures. Here we provide a brief review of the DMD
algorithm and define the types of interactions present in the simulations. We then
describe the seven models studied and discuss the importance of each. At the end
of this section, we specify the simulation parameters and the properties measured
during the simulations.
The DMD computer simulation technique was developed by Alder and
Wain-wright to calculate the properties of systems containing hard spheres or
square-well spheres.38 DMD is inherently faster than ordinary molecular dynamics, which
is based on continuous potentials, because the equations of motion for DMD can be
solved analytically. The compromise, however, is that the energy function must be
limited to discontinuous functions, such as the hard-sphere and square-well
poten-tials. Pairs of hard-sphere beads interact via the hard-sphere potential
uij(r )=
∞, r ≤σ
uij(r )=
∞, r ≤σ −, σ < r ≤λσ 0, r > λσ
(2.2)
where λσ is the well diameter and is the well depth. After the pioneering work of Alder and Wainwright, Rapaport39 and Bellemans, Orban, and Belle40 proposed a
method of simulating chain systems with discontinuous potentials wherein the bond
between neighbors along the chain is allowed to vary freely over a small range around
the true bond length. With this method, successive chain segments are essentially
decoupled, allowing the chain system to be simulated with the DMD technique.
The speed of the DMD algorithm results from the decoupled nature of the
events. Beads influence each other only when the distance between them is equal to
a point of discontinuity in their potential,e.g. a collision between hard spheres. At
all other distances, they move linearly according to Newton’s equation of motion.
Therefore, a DMD simulation is broken into a series of events, as opposed to the
series of very small time steps used in continuous-potential molecular dynamics
simulations. Nonbonded beads move freely in space and experience events at
dis-tances ofσ (a hard sphere event) andλσ (a square-well event). Bonded beads move freely over a small range,(1−δ)l≤l≤(1+δ)l, whereδis the bond tolerance andl is the ideal bond length. The choice ofδdefines the acceptable range of fluctuation in the bond length. DMD simulations proceed through time by locating the pair of
beads that will be involved in the next event, advancing all beads forward in time
to that event, and calculating the event dynamics for the event pair. This process is
performed repeatedly. Several efficiency techniques are used in this work,
includ-ing neighbor lists, binary trees, and false-positioninclud-ing, and are described in detail by
14
Seven types of chain geometry are considered in this study. The first, the
"ho-mopolymer" model shown in Figure 2.1a, is a freely-jointed, tangent, square-well
chain with 80 beads. Homopolymer models were studied previously in our group
for a range of chain lengths21,22 and serve as our base structure. The remaining
models studied are each built up from this foundation. The second model, the
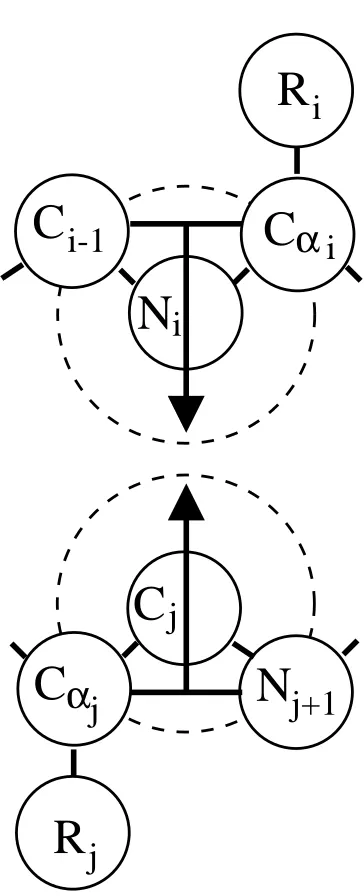
"ho-moprotein" model shown in Figure 2.1b, is a branched chain composed of a
back-bone of 60 freely-jointed, tangent, well beads and 20 freely-jointed,
square-well side-chain beads. The homoprotein geometry is reminiscent of protein models
in which each amino acid residue is represented by a four-bead monomer unit with
three beads along the backbone and one side chain bead.42–44Physically, a four-bead
amino acid representation is significantly more realistic than a one-bead
represen-tation since it allows for separate backbone and side chain interactions. The third
model, the "heteroprotein" model shown in Figure 2.1c, has the same four-bead,
branched structure as the homoprotein model but contains hard-sphere backbone
beads and square-well side-chains. In Figure 2.1, hard spheres are depicted by open
circles, and squawell spheres are depicted by filled, gray circles. The attractive
re-gions that surround square-well beads mimic attractions within real proteins, such
as those due to hydrophobic forces. Heterogeneity is an important feature in
pro-teins. The heteroprotein model, in which the side chains are attractive beads and
the backbone beads are hard spheres, is a simple representation of a peptide chain
composed of identical hydrophobic residues.
The fourth and fifth models, the “rigid homoprotein” model and the “rigid
het-eroprotein” model, are shown in Figures 2.1d and 2.1e, respectively. The rigid
ho-moprotein model (Figure 2.1d) has the same branched, square-well structure as the
homoprotein model (Figure 2.1b); the rigid heteroprotein model (Figure 2.1e) has the
same branched, mixed hard-sphere and square-well structure as the heteroprotein
model (Figure 2.1c). In each rigid model, however, extra bonds (bold, black lines
in Figure 2.1) have been added to implement rigid, angular constraints like those
present in real proteins. For the purpose of the simulation, these extra bonds are
between central atoms of neighboring amino acids are relatively fixed due to the
fixed backbone bond angles and the trans nature of peptide (inter-residue) bonds.
Finally, the side chain bead is bonded to all three backbone beads in its monomer
unit instead of just to the central bead. These extra side chain bonds are necessary
because real amino acids have a chiral center atom. Two of the chiral atom’s
con-stituents are the neighboring backbone beads; the other two are a side chain (one
of twenty different chemical groups, the side chain mimicked here) and a hydrogen
atom. In all models here, we neglect the hydrogen atom side chain. We model the
chirality of the central atom by fixing the remaining side chain bead’s position such
that the model chain is composed of identical isomers.
The final two models, the “overlapping-rigid homoprotein” model and the
“overlapping-rigid heteroprotein” model, are shown in Figures 2.1f and 2.1g,
respec-tively. The overlapping-rigid homoprotein model (Figure 2.1f) has the same
geome-try and set of bonds as the rigid homoprotein model (Figure 2.1d); the
overlapping-rigid heteroprotein model (Figure 2.1g) has the same geometry and set of bonds as
the rigid heteroprotein model (Figure 2.1e). However, the overlapping models have
enlarged bead diameters. The resulting chains have smoother overall surfaces that
more accurately reflect the atomic-level surface topography of real molecules.
We perform DMD simulations on each of the seven protein models described
above. Simulations are performed on DEC Alpha workstations and range in length
from 50 million to 1 billion collisions, with longer simulations required to reach
equilibrium at lower temperatures. For each simulation, initial chain conformations
and velocities are random. In all models, the backbone bond lengths are chosen
to be unity and all other lengths are scaled relative to the backbone bond length.
The side chain bond length, present in models (b) through (g), is chosen to be larger
than the backbone bond length to mimic the geometry of real amino acids in which
16
much larger than that of a backbone bond; the side chain bond length is arbitrarily
set to 1.30. The bond lengths and angles for models (d) through (g) are shown
schematically in Figure 2.2 and include next neighbor bond lengths of 1.50, chiral
center to chiral center bond lengths of 2.35, and side chain to non-chiral-center
backbone bead bond lengths of 1.92. For models (a) through (e), the bead diameters
(σ) are unity; for overlapping models (f) and (g), the bead diameters are 1.50. All square-well beads have well widths (λσ) of 1.5σ. The bond tolerance, δ, is set to 0.1; therefore, all bonds are held to within 10% of their ideal lengths.
During the simulations, we monitor several thermodynamic and structural
prop-erties including radius of gyration, specific heat, and internal energy. The reduced
squared radius of gyration,R∗
g, is given by
R∗
g =
D1
N
PN i=1
(xi−xc)2+(yi−yc)2+(zi−zc)2E
N (2.3)
whereNis the number of beads;xi,yi,ziare the coordinates of beadi; andxc,yc,
zc are the center of mass coordinates of the chain. The reduced specific heat,Cv∗, is
given by
C∗
v = Ckv B =
hE2−(hEi)2i
(T∗)2 (2.4)
wherekB is Boltzmann’s constant andT∗is the reduced temperature, defined to be
kBT /. The reduced internal energy,E∗, is given by
E∗= hEi
. (2.5)
Thermodynamic transitions can be characterized by changes inR∗
g,Cv∗, andE∗with
temperature. A sigmoidal shape in anR∗
g versusT∗plot is characteristic of a
second-order, gas-to-liquid collapse transition. The number of plateaus and peaks in aC∗
v
versus T∗ plot corresponds to the number of equilibrium transitions.
Disconti-nuities in an E∗ versus T∗ plot are evidence of a first-order, liquid-to-solid phase
To study the effect of branching, we compare the behavior of an unbranched
chain (the homopolymer in Figure 2.1a) with that observed for a chain with separate
backbone and side chain beads (the homoprotein in Figure 2.1b). Figures 2.3 and
2.4 show R∗
g, Cv∗, and E∗ versus T∗ for the homopolymer and the homoprotein,
respectively. In this and in all thermodynamic data shown in this paper, the symbols
represent the average equilibrium value from at least three independent simulations.
Error bars are shown on all points and are the standard deviation in the measured
values. However, the error bars are only visible when they exceed the size of the
symbol. The dashed lines on theR∗
g plots are meant only to guide the eye between
the points. The solid lines on the C∗
v and E∗ plots are the result of a weighted
histogram analysis wherein degeneracy factors are extracted from the results of
runs at different temperatures and a partition function is obtained.45 The error in
the C∗
v measurement can be quite large, especially at low temperatures, since it
is a measure of fluctuations about a simulation variable (energy). As a result, the
error bars on theCv data can be large and, at times, the histogram data lies outside these error bars. Consequently, we confirm the presence of a phase transition by
comparing pairs of plots, either a sigmoidal collapse in theR∗
g versusT∗curveand
a peak inC∗
v at the same temperature or a discontinuity in theE∗ versusT∗curve
anda peak inC∗
v at the same temperature, and by examining the chain structure at
the given temperature.
The homopolymer model exhibits multiple phase transitions, as was seen
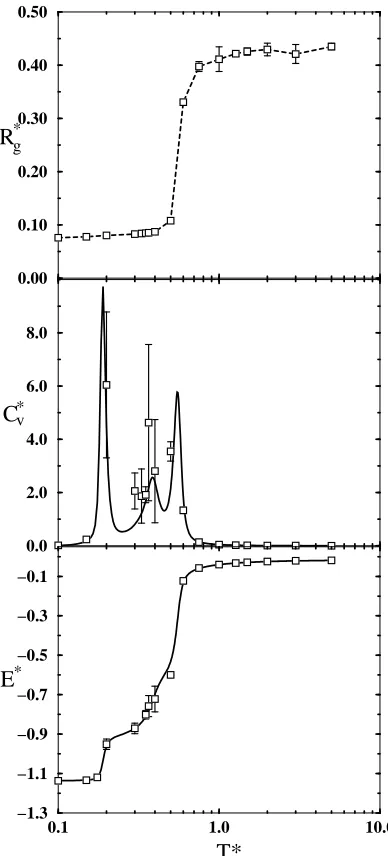
pre-viously for shorter chains.21,22 As shown in Figure 2.3, at a high temperature (T∗
of approximately 2.6 as measured at the midpoint of the transition), the
homopoly-mer system undergoes a collapse, where the R∗
g drops rapidly and the Cv∗ curve
plateaus. At a lower temperature,T∗ of 0.34, we observe a discontinuity in theE∗
plot and a corresponding spike in theC∗
v plot, behavior characteristic of a first-order
grad-18
ually, and a spike in the specific heat at approximately T∗=0.15 suggests a third
transition. However, considerable variability in Cv data at such low temperatures
makes confirming a third transition difficult. The equilibrium structures of chains
in this low-temperature regime will be discussed below. The homoprotein system
(Figure 2.4) displays almost exactly the same transition pattern as the homopolymer
system, with a collapse transition atT∗=2.6 and a lower-temperature transition at
0.34. Below T∗=0.34 the energy continues to decrease, and an increase in Cv at a
T∗of approximately 0.15 may indicate a third transition. Transition temperatures
for all models are summarized in Table 2.1. Since the only difference between these
two models is the way in which the beads are connected (each system contains 80
square-well beads and 79 bonds), it is apparent that varying the connective
arrange-ment of beads has little effect on the transition temperatures or on the strength
(steepness) of each transition. These results are in agreement with a lattice study
of branched polymers which shows that a polymer with short side chains (single
beads) adopts a global shape that is similar to that of a linear chain.46
Table 2.1: Transition temperatures observed for each model studied. Superscripta denotes rough estimates of temperature.
Transition
Model Temperatures
(reduced units)
homopolymer 2.6, 0.34, 0.15a
(Figure 2.1a)
homoprotein 2.6, 0.34, 0.15a
(Figure 2.1b)
heteroprotein 0.55, 0.19
(Figure 2.1c)
rigid homoprotein 2.5, 0.16, 0.12a
(Figure 2.1d)
rigid heteroprotein 0.50, 0.17
(Figure 2.1e)
overlapping-rigid homoprotein 3.2, 0.25, 0.15a (Figure 2.1f)
overlapping-rigid heteroprotein 0.65, 0.17 (Figure 2.1g)
homopoly-lattice geometry. The mixed-homopoly-lattice structure results from the square-well diameter
of 1.5σ at which cubic and hexagonal lattice geometries are equally stable. At tem-peratures below approximately 0.15, the chains exhibit less-symmetrical structures,
such as the one shown in Figure 2.7 for the homoprotein atT∗=0.125, in which the
simple lattice structure seen atT∗=0.30 is sacrificed for a more spherical, lower
en-ergy structure. This reorganization at very low temperatures, along with the peak in
Cv at T∗=0.15, suggests a polymorphic solid-solid transition. Similar results were
obtained previously with shorter homopolymer models.21,22
The homoprotein model, in which we introduce branching to the
homopoly-mer model, will be useful in future, more-realistic protein models because of the
increased geometric detail possible due to its discretized backbone and side chains.
As shown by the results in this section, the introduction of branching did not disrupt
the multiple thermodynamic phase transition behavior seen with homopolymers.
This behavior is qualitatively characteristic of phase transitions during protein
fold-ing and, consequently, an important behavior in any protein model.
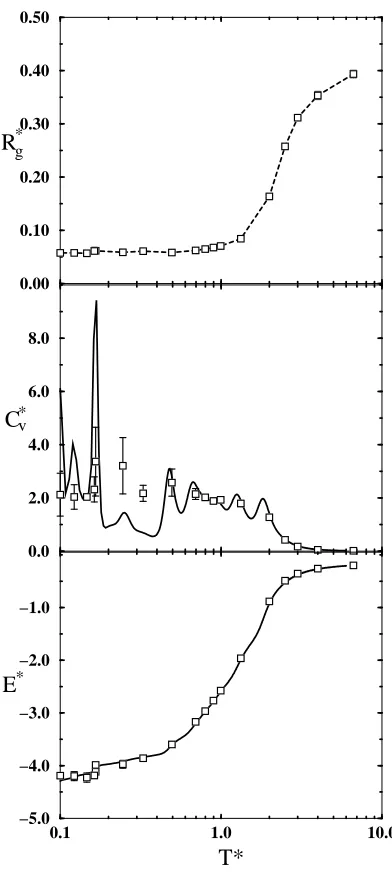
2.3.2 Chain Heterogeneity
We study the effect of heterogeneity by comparing behavior of the homoprotein,
Figure 2.1b, with that of the heteroprotein, Figure 2.1c. Figure 2.8 showsR∗
g,Cv∗, and
E∗ versus T∗ for the heteroprotein. While the same phase transitions are present
as in the homoprotein system (Figure 2.4), introducing heterogeneity causes both a
downward temperature shift and a strengthening (steeper curve) of the individual
transitions. The collapse occurs atT∗ of 0.55, and a first-order transition occurs at
approximately 0.19. (Compare with homoprotein results of T∗=2.6 and 0.34.)
Ad-ditional low temperature transitions for this model, if possible, exist belowT∗=0.1,
20
homoprotein and the heteroprotein models is expected when examined in light of
the results for a cluster (no bonds) of square-well beads.22A comparison of a 64-bead
square-well chain with a 64-bead square-well cluster demonstrates that the lack of
bonds in the cluster (a lack of physical constraints) leads to sharper, steeper curves,
corresponding to stronger physical transitions at lower temperatures. Since the
clus-ter has considerably fewer constraints than the chain, it is necessary to subject it to
a lower temperature to force a transition. In this study, the heteroprotein has fewer
constraints than the homoprotein in that the beads experiencing the attractions that
enable the phase transitions (the square-well side chains) are separated from each
other by five bonds. The system can be thought of as 20 square-well beads that are
distantly constrained to each other through loops of four hard-sphere beads. The
collapse transition occurs via the side chain square-well interactions in spite of the
fact that the side chains must drag the hard-sphere backbone along with them.
Figure 2.9 shows a symmetrical, low-temperature (T∗=0.17) conformation for
the heteroprotein chain. Since the backbone beads are not square-well, they do not
participate in ordering. The size of the backbone beads in Figure 2.9 is significantly
reduced to highlight the hexagonal ordering of the side chain beads. As in the
homopolymer and homoprotein systems, decreasing the temperature leads to even
lower-energy structures (not shown) that do not exhibit lattice symmetry.
Heterogeneity, like branching, is introduced to this hierarchy of models since
it will be beneficial in future, more-realistic protein models. The addition of
het-erogeneity allows the study of hydrophobic core formation, an important aspect of
protein folding. The low-temperature conformations observed, like the one shown
in Figure 2.9, readily adopt structures with the hydrophobic side chains clustered at
the center and the non-hydrophobic backbone beads buffering them from solvent.
However, in contrast to real protein native states, the observed structures are not
tein model, Figure 2.1d. Figure 2.10 showsR∗
g, Cv∗, andE∗ versus T∗ for the rigid
homoprotein. The rigid homoprotein system displays multiple phase transitions
with a collapse transition at aT∗of 2.5 and a first-order transition at a T∗of 0.16.
A lower-temperature transition is suggested by the peak in the weighted histogram
Cv data at a T∗ of approximately 0.12; however there is too much variability in
Cvdata at such low temperatures to confirm this third transition. These transitions
occur at lower temperatures than with the homoprotein model (compare with
homo-protein results of T∗=2.6, 0.34, and 0.15), demonstrating that lower temperatures
are required to force the rigid homoprotein chain into compact conformations. The
extra bonds introduced in the rigid model effectively work against compaction by
holding next neighbor and, in some cases, next-next neighbor beads apart. Although
the rigid homoprotein can collapse to a similar overallR∗
g as the homoprotein, it is
unable to make the subtle structural rearrangements necessary to achieve similar
low energies. At the lowest temperature studied (T∗=0.1), the homoprotein has an
E∗ of -5.25 and the rigid homoprotein has an E∗ of -4.25. This difference can be
attributed to decreased conformational freedom due to the increased number of
bonds in the chain.
We note that our downward shift in transition temperatures with increased
model stiffness is contrary to the upward shift reported elsewhere for lattice47and
off-lattice48–50 Monte Carlo simulations of stiff square-well and stiff Lennard-Jones
homopolymer chains. The difference is most likely due to significant differences
between the model chains studied. Other reports have focused on the phase
transi-tions in homopolymer chains withuniform stiffness, where stiffness is introduced
as an attraction between each bead,i, and its next-neighbor bead,i+2. In the mod-els developed here, a non-uniform rigidity results from the three types of bonds
22
Figure 2.2). The first type of bond, a constraint between each bead, i, and its next-neighbor bead, i +2, generates a rigidity similar to that introduced in the other studies and causes the chain to maintain a zigzag shape. Figure 2.11 shows the
ge-ometric constraints resulting from the other two types of bonds. The fixed distance
between consecutive central beads of each 4-bead monomer building block causes
that pair of beads and the two intervening backbone beads to remain fixed in a plane,
as shown by the bold parallelogram in Figure 2.11. In the final type of constraint,
side chain beads are pinned to all three backbone beads of the monomer unit such
that each monomer is fixed in a pyramid shape, as shown with bold lines on the
right in Figure 2.11. The consequence of this non-uniform rigidity is that rotational
motion around different bonds is hindered to different degrees, and the hindered
motion opposes ordering of the chain.
The structures observed for the rigid homoprotein model reflect the influence
of the extra geometric constraints depicted in Figure 2.1d. With the homoprotein
model, the collapse transition always results in spherically-shaped structures.
How-ever, in rigid homoprotein runs, collapse of the chain results in either spherical or
ellipsoidal conformations, such as the ellipsoidal structure shown in Figure 2.12a
(front view) and b (side view) for a T∗ of 0.15. While the packing appears quite
dense, lower-energy ordered structures, such as the one shown in Figure 2.12c for
aT∗of 0.10, are possible. The extra bonds present in the rigid homoprotein model
prevent the chain from adopting the cubic- and hexagonal-packed structures like
those observed for the homoprotein chain. Ellipsoidal structures similar to those
seen here were shown previously to be characteristic of semiflexible polymers.50,51
We also consider the effect of chain rigidity on phase transitions for
heteroge-neous chains by comparing the results from the heteroprotein model, Figure 2.1c,
with those from the rigid heteroprotein model, Figure 2.1e. Figure 2.13 showsR∗
g,
C∗
v, and E∗ versus T∗ for the rigid heteroprotein. The rigid heteroprotein system
displays multiple phase transitions with a collapse transition at aT∗ of 0.50 and a
first-order transition at aT∗of 0.17. As in the comparison above for homogeneous
temper-heteroprotein is able to achieve equally low energies as the temper-heteroprotein (compare
Figure 2.8 and 2.13). This can be explained by the fact that the side chains alone
account for the energy of the system. The extra bonds in the rigid heteroprotein
model make overall compaction more difficult; however, once the side chains are
clustered in the center of the structure, they are able to make the rearrangements
necessary to achieve low energy states.
The structures for the rigid heteroprotein model, such as the one shown in
Fig-ure 2.14 for T∗=0.16, are notably similar to those observed for the heteroprotein
(compare with Figure 2.9). Again, it is the side chains that dictate structure in these
heterogeneous models; the backbone beads are not involved in stabilizing
equilib-rium structures. Once the side chain beads have aggregated in the center of the
conformation, the pseudobonds have little impact and do not prevent the
forma-tion of structures with local ordering of the side chains.
In a real amino acid residue, the backbone bond angles are fixed and the side
chain is constrained to a particular location relative to the backbone atoms. These
constraints severely limit the conformational freedom of the atoms in the amino
acid residue and contribute to important features of folded protein structure, such
as α-helices andβ-sheets. A protein model that utilizes multiple backbone beads to represent a single amino acid residue should incorporate constraints that mimic
the constraints in real amino acid residues. The rigid homoprotein and rigid
het-eroprotein models here include these important constraints in the form of extra
bonds between next-neighbor beads and, in some cases, next-next-neighbor beads.
The simulation results show that the rigid models (Figures 2.1d-e), despite their
decreased flexibility when compared with the non-rigid models (Figures 2.1b-c),
ex-perience multiple thermodynamic phase transitions which qualitatively correspond
to the stages of protein folding and successfully bury hydrophobic side chains, two
24
2.3.4 Overlapping Beads
To study the effect of bead overlap, we perform simulations of the
overlapping-rigid homo- and heteroprotein models shown in Figures 2.1f and g, respectively.
These models have bead sizes that are 1.5 times larger than in the previous five
model systems, as depicted in Figures 2.1 and 2.2. Figure 2.15 shows R∗
g, Cv∗, and
E∗versusT∗for the overlapping-rigid homoprotein. This system displays multiple
phase transitions with a collapse transition at a T∗ of approximately 3.2, a
first-order transition at aT∗of 0.25, and a lower-temperature transition as suggested by
the CV data at T∗ approximately equal to 0.15. These transitions occur at higher
temperatures than for the rigid homoprotein model (T∗=2.5, 0.16, and 0.12). The
shift in transition temperatures and the ability to reach lower overall energies
re-sult directly from the greater range of attraction for each square-well bead in the
overlapping-rigid homoprotein model.
The structures observed for the overlapping-rigid homoprotein model are
sim-ilar to those for the rigid homoprotein model and reflect the influence of the
next-neighbor and next-next next-neighbor bonds depicted in Figure 2.1f. In some
overlapping-rigid homoprotein runs, collapse of the chain results in rod-shaped conformations,
such as the one shown in Figure 2.16a (side view) and b (top view) for aT∗of 0.23.
Rod-like structures are shown elsewhere to be characteristic of semiflexible
poly-mers.50,51 Other low-temperature structures, such as the one shown in Figure 2.16c
for a T∗of 0.10 tend to be ordered and spherical. The extra bonds present in the
overlapping-rigid homoprotein model hinder the flexibility of the chain and prevent
it from adopting structures with cubic- and hexagonal-packing similar to those
ob-served for the simple homoprotein chain.
Figure 2.17 showsR∗
g,Cv∗, and E∗ versus T∗ for the overlapping-rigid
hetero-protein model shown in Figure 2.1g. The overlapping-rigid heterohetero-protein system
displays multiple phase transitions with a collapse transition at aT∗ of 0.65 and a
first-order transition at a T∗ of 0.17. The collapse occurs at a higher temperature
from the small number of hydrophobic beads in these systems (twenty side chains)
and the heterogeneity of the chain. After the collapse transition, the side chains
(pale beads) are clustered in the center of the structure and the backbone (dark
beads) snakes around the outside. Subsequent, lower-temperature transitions
in-volve rearrangements of these relatively-uncoupled side chain beads. Once the side
chains have been segregated and tightly packed in the core of the structure, small
changes in the range of the attractive wells are relatively unimportant. In contrast,
the comparison above between the rigid homoprotein and the overlapping-rigid
ho-moprotein showed a shift in the low-temperature transition. In the homogeneous
systems, all beads have attractive wells and small changes in structure can have a
dramatic effect on the overall energy depending on the range of the attractive well.
We observe similar side-chain packing geometries for all heteroprotein models.
As with the heteroprotein and rigid heteroprotein models, the overlapping-rigid
het-eroprotein model displays symmetrical side-chain conformations, such as the one
shown in Figure 2.18 for a T∗ of 0.16. Figure 2.18a shows the chain with reduced
bead diameters to highlight the symmetrical arrangement of side chain beads.
Fig-ure 2.18b shows the same structFig-ure from a different angle with full-size beads to
demonstrate the way in which the backbone snakes along the outer surface of the
structure, encasing the hydrophobic side chains.
Real protein atoms have significant overlaps. Spacefilling pictures of proteins
at the atomic level look more like the overlapping models introduced in this section
than the five tangent-bead models discussed previously. To properly mimic real
pro-tein steric interactions, appropriate bead size to bond length ratios should be chosen
when constructing a protein model. The results shown here indicate that
overlap-ping chains are capable of displaying protein-like character. The overlapoverlap-ping-rigid
models are able to selectively bury hydrophobic side chains and experience