Copyright 1974 American SocietyforMicrobiology Printed in U.S.A.
Isolation and
Partial Characterization
of
Different
Defective
DNA
Molecules Derived
from
Polyoma
Virus
MIKE FRIED
Imperial Cancer Research Fund, Lincoln's Inn Fields, London WC2A 3PX, England
Received forpublication 19 November 1973
Supercoiled DNA molecules purifiedfrommousecellsinfected with
high-mul-tiplicity-passaged polyoma virus has a broader size distribution and sediments
moreslowly than DNA derived fromlow-multiplicity-passagedvirus. The shorter
DNA moleculesare predominately noninfectious. Virus populations containing
distinct size classes of defective virus DNAwere isolated by growing virusfrom
single cells infected by a defective and nondefective helper virus (infectious
center). This technique probably results in the cloning of defective virus
particles. By applying the infectious center method to DNA from various
fractions ofsucrosegradients it has beenpossibletoobtainshorter circular DNA
molecules ranging in size from 50 to 95% of the unit-length polyoma DNA
molecule. The shorter molecules inanyonepreparationarehomogeneous in size.
This class size is retained upon repeated passage of crude viral lysates at high
multiplicity. Thus far, all thepurified shorter DNA molecules testedappeartobe
noninfectious and largely resistant to cleavage by the RI restriction enzyme.
Someof the purified defective molecules have been found tointerfere with the
production of infectious virus upon co-infection with unit-length infectious
polyomaDNA.
Heterogeneity in the size of both polyoma and
SV-40 viral DNA extracted from viruspassedat
high-input multiplicities has been observed by
a number of workers (1, 7-9, 11-13).
Prepara-tions of this type contain a significant number
of shorter molecules that are predominately
noninfectious (1, 9, 11-13). If these shorter
moleculesrepresentpartially deleted virus
mol-ecules, they couldproveextremely useful in the
mapping and identification of viralgenes,
espe-cially those involved in neoplastic
transforma-tion. The following report describes the
isola-tion and partial characterization of different
polyoma virus preparations greatly enriched in
shorter supercoiled DNA molecules that range
in size from 50 to 95% of the unit-length
molecule.
MATERIALS AND METHODS
Virus stocks. The wild-type large plaque (PY)
strain and the temperature-sensitive mutant TS-A
havebeen described elsewhere (3). Virus stockswere
grown on mouse embryo cells, and the virus was
liberated from theinfected cells by freeze-thawingor
sonicationinthepresenceof thegrowthmedia(4),or
both.
Some of the properties of the high-multiplicity,
low-multiplicity, and starting virus (medium
multi-plicity) aresummarizedinTable 1.
The initial starting virus used to produce the
high-multiplicity virus had been passed withnogreat
care over a number ofyears. The virus was passed
undiluted foranumberofpassages,usually infecting5
[image:1.494.47.444.570.660.2]x 106 mouse embryo cells with 0.5 ml of lysate. High-multiplicity virus is denoted by DEL followed bythepassagenumber. For instance, the fifthpassage
TABLE 1. Characteristics of virus produced at differentmultiplicities
Characteristic
~~Low-
Medium-High-Characteristic | multiplicityvirus multiplicityvirus multiplicity virus
PFU/HAU ... 106 105 As low as 103
PFU/physical particle... 1/20 1/200 Aslowas 1/20,000
DNAPFU/ggofDNA ... 106 105 As low as 103
Yield of PFU/2 x 106 cellsa (Single
cycle) ... 8 x 109 8 x10" As low as 8 x 106
YieldofDNA/5 x 106cellsa...5-8....58g 2-4gg As low as 0.5-1 tg
aInput multiplicity of 5 PFU/cell.
9:39
on November 10, 2019 by guest
http://jvi.asm.org/
of thewild-type virus (PY) is referred to as PY-DEL-5
and the sixth passageofTS-AasTS-A-DEL-6.
Low-multiplicity virus wasderived from the
medi-um-multiplicity virus by isolating single plaques from dishes containing less than four plaques per dish. The plaques were picked into 1 to 1.5 ml of growth medium
and freeze-thawed three times. Each plaque usually
contained between 104 and 106 PFU. Approximately
onehalf of the virus from the plaque was used to infect
5 x 106 cells. The virus from this infection was
harvested after5 to 7daysat 37Cor10 to14days at
31.5 C. "4C-labeled viral DNA purified from cells
infected with low-multiplicity virus at an input of 1
PFU/cellwasusedasthestandard unit-length marker
DNA insucrosegradients.
Assayof virus and infectious DNA. The plaque
assay usedto titer infectious virus and the
haemag-glutination assay used to titerphysical particles have
been described previously (4). Infectious viral DNA
wasassayed on mouse embryo cells by the
DEAE-dex-tranmethod ofThorne et al. (9).
Infectious center plaques. The cloning of
individ-ual defective virus particles from
high-multiplicity-passaged viruswas attempted by growing virus from
single cells infected by adefective and a nondefective
particle. Mouse embryo cells were infected at a
multiplicity of less than, or equal to, 1 PFU/cell, but
at amultiplicity of greater than one particle per cell.
The inputmultiplicity dependedonthe defectiveness
ofthe stock asestimated from the
PFU/hemaggluti-nation unit (HAU) ratio. Usually a variety of input
multiplicities wereusedforeach viruspreparation.
After virus absorption the cells werewashed three
timeswith medium and then incubatedfor2 to 3 hin
the presenceofreceptor-destroying enzyme and
anti-polyoma antiserum to reduce and inactivate the
unabsorbed virus (4). The infected cells were then
trypsinized and reseeded at concentrations ranging
from50to5,000cells with2 x 106uninfectedmouse
embryocells per 50-mm petri dish. After2to 3h when
the cells had settled, the media were removed and
agarwas addedasforanormal plaqueassay.
When the infectiouscenterplaques developed they
were picked into 1 to 1.5 ml of medium with a
wide-bore pipette. The picked plaques were then
freeze-thawedthree timestorelease the virus from the
plug of agar. Thevirus wasthen grownup on mouse
embryo cells at high multiplicity. The supercoiled
DNA derivedfromvirusproduced bythese infectious
center plaques was then sedimented in neutral
su-crosegradients.
Viral DNA extraction and purification of
su-percoiled molecules. Infected mouse embryo cells
wereincubatedforeither4daysat37Cor7to8days
at 32C(usuallyinthepresenceof2MCiof [3H
Ithymi-dine and 1 ug of cold thymidine of medium per
milliliter) before viral DNA was extracted by the
selective method of Hirt (5). The Hirt supernatant
fluidwasextracted withphenol,and the nucleicacid
was precipitated by the addition of 2 volumes of
ethanol at -20C. The supercoiled and
nonsuper-coiled DNA were separated by using an ethidium
bromide-cesium chlorideequilibrium gradient (6).
Neutral sucrose gradients. Linear 5to20% (wt/
vol) sucrose density gradients containing 0.14 M
NaCl, 0.01 MTris-hydrochloride (pH 8.5), and 0.004
M EDTA were used for sedimentation analysis of
purified supercoiled DNA. [3H]thymidine-labeled
DNAsamples with or without [14C]thymidine-labeled
marker DNA involumes of 0.2 to 0.3 ml were layered
on topofgradients that were centrifuged in anSW40
rotor at 28,000 rpm for 17 h at 19 C.The tubes were
pierced from the bottom, and either 5 or 10 drop
fractions werecollected on 12-inch (2.542 cm)
What-man no. 1 filter paper. There were 64 to 67 10-drop
fractionsor128 to 1345-drop fractions. The DNA was
acid precipitated and the radioactivity determined.
Thesizeof slowersedimenting peaks, expressed as a
percentage of the unit-length marker DNA, was
calculated from the equationofVinograd (2).
Cleavage withRIendonuclease. The R,
endonu-clease was a gift ofW. R. Folk. The digestion was
carried out in a buffer containing 0.05 M
Tris-chlo-ride, 0.05 M NaCl, and0.01M MgCl2at 37C for 30
min. Theenzyme wasin20-fold excess at a ratio of 1
Lliter ofenzyme to 1 ,g of DNA. The reaction was
stopped by the addition of EDTA to a final
concentra-tionof 0.03 M.
RESULTS
Isolation of TS-A-shorter DNA molecules.
It wasthought that a TS-A viral DNA deleted
in genes coding for capsid proteins could be
rescued by a temperature shift of mouse cells transformed and maintained atthe
nonpermis-sive temperature (10). Therefore, the isolation
of shorter DNA molecules from TS-A was
at-tempted.
Upon repeated passage of TS-A virus at
high-input multiplicity, it was observed that the yield of particles as well as the yield of
plaque-forming units decreased markedly. At
the sixth high-multiplicity passage the
PGU-HAU ratio was reduced to 2 x 103, which was
50-fold less than the starting virus inoculum and 500-fold less than low-multiplicity-pro-duced virus. This is equivalent to about 1
PFU/10,000 physical particles (usually about 50% ofthese particles do not contain any viral DNA). The major part ofthesupercoiledDNA extractedfromTS-A-DEL-6-infectedcells sedi-mented more slowly than low-multiplicity marker unit-length DNA in neutral sucrose
gradients.Acomparisonofthe widths of thetwo
sedimenting bands (Fig. 1) indicates that the
TS-A-DEL-6 DNA is moreheterogenous in size
than the low-multiplicity marker DNA.
To see ifcomplementation to form a plaque
between different defective virus particles
pres-ent in the TS-A-DEL-6virus population could
be detected, 43 infectious center plaques were
isolated after infection with TS-A-DEL-6 virus at a multiplicity of 0.01
PFU/cell
(approxi-mately 100physicalparticles/cell).
All43on November 10, 2019 by guest
http://jvi.asm.org/
E
CL
Fraction Number
FIG. 1. Neutral sucrose gradient sedimentation
analvsis of TS-A-DEL-6 DNA. Symbols: 0,
3H-labeled TS-A-DEL-6 DNA; 0, 14C-labeled
unit-length marker DNA.Inthis and thefollowing figures
only data from the lowerhalf of thesucrosegradients
arepresented.
tious center plaques when replaqued showed
linear plaque formation with dilution. This
suggests that these plaques were not produced
by complementation of two or more defective
virusparticles. All theplaquestested contained
between 104and 106 PFU.
To isolate shorter DNA molecules, seven
infectiouscenterplaques derived froman
infec-tion ofTS-A-DEL-6 atan input multiplicityof
0.001 PFU/cell (approximately 10physical
par-ticles/cell) weregrownupasdescribed in
Mate-rial and Methods. Theresulting virus wasused
to infecta large numberofmouseembryocells
fromwhich supercoiled DNAwasextracted and
purified.
The DNA from each of theseven plaqueswas
sedimented ina neutralsucrose gradientinthe
presenceofamarker fromlow-multiplicity viral
DNA. The width of each oftheseven
DNA-sedi-menting bands indicated that they were more
homogeneous in size than the DNA from the
uncloned TS-A-DEL-6 DNA. DNA from three
ofthe plaques sedimented coincident with the
marker, whereas two sedimented with
charac-teristics ofmolecules 5 to 10% shorter and two
approximately 15% shorter.
The infectivity of the fractionated DNA of
one oftheplaques (TS-A-DEL-6-2) containing
15% shorter molecules was assayed (Fig. 2). It was found that the peak of infectivity sedi-mented faster than the bulk of theDNA, and in 4 thesame region asthe low-multiplicitymarker
DNA. In the fractions containing most of the
15% shorter DNA, only background infectivity
was present. Eighty-seven plaques resulting
from infection with DNA from thelight side of the infectivity peakin theregionofthe bulk of the DNA were picked. All of these plaques contained 101 to 106 PFU and, on replaquing, produced alinear dose response withdilution. 2 o. Clonal isolation of PY shorter DNA
molecule. Because the shorter molecules de-rived from TS-A appeared to carry little or no
infectivity, complementation between
wild-type shorter DNA molecules and the various temperature-sensitive mutants, including TS-A, might be feasible. Therefore, the
isola-tion ofshorter DNApopulationswasattempted
from wild-type virus. After the second passage at high multiplicity of wild-type virus,
infec-tious center plaques were isolated and grown
up. Two out of three plaques tested showed a
u
c x
9
9
x
9
FRACTION NUMBER
FIG. 2. Neutral sucrose gradient sedimentation
analvsisof DNA from infectiouscenterplaque TS-A-DEL-6-2. (A) DNA infectivity of various gradient
fractions of TS-A-DEL-6-2 DNA. Symbols: 0,
3H-labeled TS-A-DEL-6-2 DNA; A, infectious DNA
plaque-forming units. (B) Sedimentation behaviorof
TS-A-DEL-6-2 DNAinrelationtounit-length marker DNA. Symbols: *, 3H-labeled TS-A-DEL-6-2 DNA;
0, "4C-labeledunit-length marker DNA.
. 4
K
'.->..
,01
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.494.44.238.58.307.2] [image:3.494.250.438.331.580.2]sharpsingle peakofDNAabout5 to 10%shorter
than the marker, whereas the DNA from the A
third plaque co-sedimented with the marker.
An attempt was made to enhance for even
shorter populations by growing up virus from
the DNA fromthelight sideofhigh-multiplicity X DNA peaks. Supercoiled DNA, purified from -PY-DEL-5 sedimented through a neutral
su-crosegradient, showedabroaddistribution and
sedimented moreslowly than the marker DNA (Fig. 3). DNA from the peak fraction and a
number of fractions from the light side of the
peak of the PY-DEL-5DNA were usedtoinfect B
mouse cells. The viruses produced from these 3
DNA infections were used to infect a large
number of mouse cells from which the
[image:4.494.265.455.63.330.2]super-coiled DNAwas purified andanalyzed. i 2
Figure 4shows the sedimentation characteris- 2
tics of DNA derived from these various frac- T
tions. It can be seen that the DNA obtained 5
from the peak fraction 14 is similar in its size
distribution to that ofthe parental PY-DEL-5
gradient (Fig.
3A). With the DNA derivedfrom ,cIl 0
the
lighter
fractions,
twopeaks
of DNA were 20 30resolved; one co-sedimented with the marker Fraction Number
DNA, whereas the other sedimented more FIG. 3. Neutral sucrose gradient sedimentation
slowly depending on its origin in the parental
analvsis
of PY-DEL-5 DNA. (A) Sedimentation ofgraien.Iwafundtha th aoun
ofsloerPY-DEL-5
DNA. The DNA of fractions 14-22 wasgradient. It was found that the amount of slower used to makevirus stocks that were furtheranalyzed
sedimentingDNA decreases muchmorequickly (see Fig. 4). (B) Sedimentation behavior of PY-DEL-5
than the unit-length DNA upon infection with in relation to unit-length marker DNA. Symbols: 0,
increasing dilutions of the virus derived from 3H-labeled PY-DEL-5 DNA; 0,
"4C-labeled
unit-thevarious fractions.Thusitwouldappearthat length marker DNA.
14, 15 6
k.
J: X~
3,'0
2-1Ao 0 2200 1 251 5 0 20 25
/1
3. 1711.
15. 130D~~~~~~~~3/3
Fractlion Number
FIG. 4. Neutral sucrose gradient sedimentation analysis of supercoiled DNA isolated from mouse cells
infected with virus derived from the DNA of gradient fractions 14-22 (Fig. 3A). Symbols: *, 3H-labeled DNA
derived from gradient fractions 14-22; 0, "4C-labeledunit-length marker DNA.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.494.116.405.412.638.2]the shorter molecules are defective and are
dependent on the unit-length molecules for their growth.
The viruses derived from fractions 21 and22
(Fig. 4) were used to produce infectious center
plaques. Fourteen infectious center plaques
were grown up and theirDNAwasanalyzed. In
all cases most ofthe DNAco-sedimented with
the marker. In most of the preparations a
shoulder or another peak of DNA was distin-guishable. Table2lists theapproximate size of the slower sedimenting DNA relative to the unit-length molecule. Figures 8A and 9A show representative gradients of preparations
con-tainingmolecules75and60% oftheunit-length molecule, respectively.
Infectivity of the shorter molecule. It was
already found that the infectious DNA derived
from TS-A-DEL-6-2 (Fig. 2) sedimented inthe region ofthe unit-length molecule rather than with the bulk of the DNA. It is ofinterest to
know if all the DNA sequences present in the unit-length molecules of polyoma virus are
essential for the production of new infectious virus. Thus it was decided to look at the infectivity ofother isolated shorter DNA mole-cule populations.
The 75% DNA molecules derived from virus
of the plaque designated 21-2 were purified through twosucrose gradients. The final DNA
preparation (Fig. 5) contained 3 x 102PFU/ug of DNA. This is equivalent to a 104-fold
de-TABLE 2. Sizes of shorter DNA moleculesofvirus
derivedfromfraction21and22
Approximate size Infectious center slower
plaques
~~~~of
slowerplaques sedimentingDNAa
Fraction21
21-1 75%
21-2 75%
21-3 50%
21-4 50%
21-5 75%
21-6 None
21-7 None
Fraction22
22-1 60%
22-2 None
22-3 75%
22-4 None
22-5 90%
22-6 75%
22-7 65%
aThe sizes of the slower sedimenting DNA,
ex-pressed as a percentage of the unit-length marker
DNA, was calculatedfrom the equation of Vinograd
(2).
[image:5.494.248.442.60.212.2]1 09 20 30 40 Fraction Number
FIG. 5. Neutral sucrose gradient sedimentation
analvsis of 75% DNA derived from virus from
infec-tious center plaque21-2. Thepeakcontinuing the 75%
DNA wasisolated from two successive neutral sucrose
gradients before being sedimented with the marker
DNA. Symbols: *, 3H-labeled 75% 21-2 DNA; 0,
"4C-labeledunit-length marker DNA.
crease in terms of plaque-forming units per
moleculesofDNAincomparison with low-mul-tiplicity viral DNA. (Table 1). Sixplaquesthat
arose fromthe 21-2 75% DNAwere pickedand
replaqued. Individualplaquesweregrown up at
low multiplicity. Supercoiled DNA obtained fromthe virus fromtheseplaqueswasanalyzed
in sucrose gradients and each plaque that
de-rived DNA showed a single peak that co-sedi-mented with the marker DNA, asillustratedin
Fig. 6. This resultsuggeststhe small amount of
infectivity present in the 75% molecules of
plaques 21-2 is probably due to contamination with unit-length molecules.
The infectivity found in 90 and 50% mole-cules, isolated aftertreatment with the
restric-tionenzyme
RI
(seebelow),wasless than15and125
PFU/4g
of DNA, respectively. All the shorter moleculesso faranalyzed are defective inthe production ofnewinfectious virus.A B
0 1 \ R
I~~ ~~~~2§i
;0-F-ct..N-mb.,F{b um
FIG. 6. Neutral sucrose gradient sedimentation analvsis of DNA derived from virus from two plaques
thataroseafterinfection with purified 75% 21-2 DNA
(Fig. 5).Symbols0, 3H-labeled DNA from 75% 21-2
plaques; 0, "4C-labeledunit-lengthmarker DNA.
I
10-1
6
I
IL
u
.T
5
c;
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.494.39.232.433.632.2] [image:5.494.249.440.493.628.2]J.VIROL.
Restriction enzyme
RI
sensitivity of theshorter molecule. Circular polyoma virus DNA
is cleaved to linear molecules at one unique
region by
RI
restriction enzyme (D. Robbersonand M.Fried, manuscriptinpreparation).Viral
DNA derivedfromplaques grown at low
multi-plicity was found to be greater than 95%
sensi-tive tocleavageto linear moleculeswith the R,
restriction enzyme (Fig. 7).
A number ofpreparations containing both a
peak of unit-length molecules and a peak of
shorter moleculesweretested for their
sensitiv-ity to cleavage with
R,.
So far none of theshorter molecules (14 isolates) were found, to
any great degree, to be sensitive to cleavage
with the R, enzyme. Unit-length molecules in
the same preparations were cleaved to linear
molecules (Fig. 8 and 9). If detectable amounts
ofthe shortermoleculesweresensitiveto cleav-age with
RI,
a disappearance of the shorter supercoiled molecules should have occurred andapeak of DNA sedimentingatthe region where
the shorter linear molecules sediment would
have been observed.
Treatment with
RI
revealed that resistant supercoiled molecules about 85 to 95% of the unit-length molecules are present in varyingamountsin most preparationsproduced athigh
multiplicity. These R,-resistant molecules are
particularlyobvious in preparations containing DNA less than 80% where they appear as a
shoulderor a separate peak (Fig.8 and 9). Treatment with
RI
was used to purify the shorter molecules. The preparations weretreated with
RI,
and the resistant shortersuper-coiled moleculeswereseparatedfromthe sensi-tive linear molecules by equilibrium density centrifugation. Velocity sedimentation was
FIG. 7. Neutral sucrose gradient sedimentation
analvsis of low-multiplicity DNA before and after
treatment with restriction enzyme RI. (A) Before
treatment withRI. (B)AftertreatmentwithRI. The marker DNAwasaddedaftertheR,reactionhadbeen
terminated.Symbols: *, 3H-labeledlow-multiplicity
DNA; 0, 14Cunit-length marker DNA.
A
F,..ti.. Number
FIG. 8. Neutral sucrose gradient sedimentation
analvsis of DNA from infectious center plaque 22-3
(Table 2) before and after treatment with restriction
enzyme R,. (A) Before treatment with RI. (B) After treatmentwithRI.The markerDNA was added after
the RI reaction had been terminated. Symbols: 0,
3H-labeled 22-3 DNA; 0, "4C-labeled unit-length
marker DNA.
6 A 6
5
~~~~~~~~~~~~~~~5
7!~41
~~~~~~~~~~~~~~~~~4
;
x~~~~~~~~~~~~~
~~~~~~~~~~~~~~~~~~~~i
u 333
2
-2
10 20
S B 6
51
5x6
4 43
13
0
12 /2
1
10
3 10 20 30
Fraction Number
FIG. 9. Neutral sucrose gradient sedimentation
analvsis ofDNA from infectious centerplaque 22-1
(Table 2) before andaftertreatmentwithrestriction
enzymeRI. (A) Before treatment with RI. (B)After
treatmentwithRI. ThemarkerDNAwasaddedafter
the RI reaction had been terminated. Symbols: 0,
3H-labeled 22-1 DNA; 0, "4C-labeled unit-length
marker DNA.
used as a further purification step for those
molecules of80% orless.
944 FRIED
I
i 0
,dj'
,
I
B |
w
2,
I
I
I, 11
I
.3
1
/II
p
i. ,i
1.
1-11
-
-Zk-E
I
.x
I
-II
on November 10, 2019 by guest
http://jvi.asm.org/
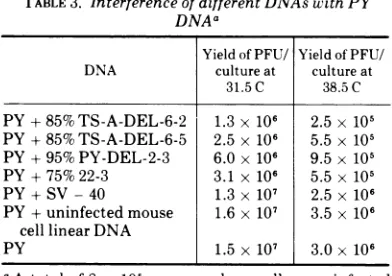
[image:6.494.269.456.64.212.2] [image:6.494.263.460.300.562.2] [image:6.494.63.253.477.603.2]Interference. Experiments were performed
to determine the effect of the various shorter molecules on the growth of
low-multiplicity-produced wild-type DNA. Mouse cells were
infected with different preparations ofpurified shorter DNA
plus
wild-type low-multiplicity DNA. Theyields of virusproduced at 31.5and38.5 C of the dual infections were compared
with the yield producedby thewild-typeDNA alone. Table 3 lists the results of such an
experiment. Whereas co-infection with circular supercoiled SV-40 DNA, or linear DNA from uninfected cells, produced littleor noinhibition
ofthe productionofwild-typevirus,someofthe preparations ofshorter DNAproducedasmuch
as a 10-fold inhibition. Those shorter molecules derivedfromTS-Avirus areequallyeffective in inhibitingatthe permissive andnonpermissive
temperature. Thisinhibitory effect will present
a problem in the use ofgenetic
complementa-tiontests toidentify viralgenes present insome
ofthe shorterDNA molecule preparations.
DISCUSSION
It has been possible by the techniques de-scribed above to produce different viral DNA
preparations that are greatly enriched in
indi-vidual distinct classes ofsupercoiled, defective DNA molecules, ranging insize from 50 to95%
oftheunit-lengthpolyoma DNA molecule. The rationaleofthe infectiouscentermethodwas to
clone defective virus particles by growing the
progeny ofindividual cells infected byonlyone
defective particle in thepresence of a
nondefec-TABLE 3. Interference of different DNAs with PY
DNAa
YieldofPFU/ YieldofPFU/ DNA cultureat cultureat
31.5C 38.5C
PY+85%TS-A-DEL-6-2 1.3 x 106 2.5 x 105
PY + 85%TS-A-DEL-6-5 2.5 x 106 5.5 x 105
PY+ 95%PY-DEL-2-3 6.0 x 106 9.5 x 105
PY+75%22-3 3.1 x 106 5.5 x 105
PY+SV-40 1.3 x107 2.5 x 106
PY+uninfectedmouse 1.6x 107 3.5 x 106
cell linear DNA
PY 1.5 x107 3.0 x 106
aAtotal of 8 x 105 mouse embryocellswere infected with 0.25Agofunit-length (PY) DNA in the presence
orabsenceof 0.05mgofeach DNA to be tested. After
infection thecells were trysinized and 4 x 105 cells
wereplatedinto each oftwo 35-mm petri dishes with
medium containing receptor-destroying enzyme (4).
One half ofthe dishes were incubated at 38.5 C and
theother halfat 31.5 C. After 68 h at 38.5 C or 144 h
at 31.5C, the virus was harvested. The yield of
plaque-formingunits per culture is shown.
tive helpervirus particle. The observation that
30 to60% of theinfectious center derived virus
isolates contained no detectable shorter
mole-cules suggests that cells that produced shorter
molecules were infected by only one (or just a
few) defective particles. The success of this
method in the cloning of individual particlesis
supported by thefollowing observations. (i)The
shorter molecules inany oneisolateare
homoge-neous in sizeand this size molecule isretained
upon repeated passage of crude viral lysatesat
high input multiplicity. (ii) Some of the
puri-fied shorter molecules have been shown to be
homogenous with respect to their nucleic acid
sequences by heteroduplex studies (Robberson
and Fried, submitted for publication) and by the production of molar equivalents of
frag-mentsaftertreatment with multicut restriction
enzymes (Griffin and Fried, manuscript in
preparation). (iii) The shorter molecules are
representative of the size distribution of the
DNA population ofthe virus used to produce the infectious centers (Fig. 4 and Table 2).
The finding that shorter molecules retain their sizewhen grown with different isolatesof
unit-length infectious DNA molecules (Fried, unpublished data) suggests that the shorter moleculesareactually derivedfromtheoriginal population as opposed to being continuously generated by particular unit-length molecules. The infectiouscentermethod requiresonly that the defective particlebeing isolatedcan coexist
with the infectious unit-length molecule inthe formation of a plaque and thus is nonselective
to thisextent.
In most high-multiplicity-passaged virus
stocks, 85 to 95% length DNA makes up the
majority of the non-unitlengthDNAmolecules
(see Fig. 1 and 3). This may explain the
presenceof85 to95%molecules, as a second size
class, in some of the defective DNA isolates. This second size class was usually observed in
isolates containing defective DNA of less than
80% unit length (Fig. 8 and 9). The defective
DNAmoleculesso far studied have been
nonin-fectious and have depended, forcontinued
pas-sage, on complementation with unit-length
in-fectious viral DNA. The shorter DNA is thus
itself maintained by passage of virus stocks at
high multiplicity. This passage has probably
led, insome cases, to the generation of the 85 to
95% shorter molecules discussed above. The
prevalence ofthe 85 to 95% molecules may be
explained by the less efficient encapsidation
into astable particle of smaller than 85% DNA,
thus conferring a selective advantage to these
newly generated larger defective molecules. It
cannot be ruled out, however, that the second
VOL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.494.38.234.443.581.2]class of defective DNA is due to infection by
morethanonedefective particleofthe cell that
producedthe infectious center.
So far all the shorter molecules tested have been found to be predominately noninfectious
andresistant tocleavage with the
RI
restrictionenzyme. Thereasonforthisresistance to
cleav-age by
RI
is unclear at this time, but may be revealed when the structure ofthese molecules is determined. The loss ofthe R, cleavage sitemight indicate that this region is not very
essential for viral growthor thatthe defectives
aregeneratedbya common mechanism. The finding that some of the shorter mole-cules interfere with the production of infectious virus is not too surprising. On continued high-multiplicity passage both the yields of
infec-tious virus and virus particles decrease
mark-edly. Continued high-multiplicity passagealso results in the production of predominately
shorter than unit-length supercoiled DNA.
Per-haps thistypeof interferenceplayssomerolein
vivo. Defective viruses, whether due to an
addition of host sequences or repetitions of
specific viral sequences, may have a selective advantage and replicate faster and at the
ex-pense ofthe infectious virus.Suchamechanism
would lead to the slowing down of the virus infection within a high-multiplicity virus area,
suchas alesion, and might allowmoretime for
the host defense mechanism, like the immune
response, to act before the virus infection has
damagedtoomany cells.
Thetransforming abilityofdifferent purified defective DNA is now under investigation. By
this means it maybe possibleto identify those
portions of the viral genome necessary for
neo-plastic transformation, as well as other viral functions.
Some of the classes of molecules isolated contain only a portion of the viral genome
(Robberson and Fried, manuscript in
prepara-tion). Thus, the isolation procedures described above present a way of obtaining large and probably homogeneous quantitiesofarestricted portion of this small viral genome that can be
replicated in vivo.The biological, biochemical,
and genetic studies ofthese deleted molecules
should lead to a better understanding of the properties of polyoma virus.
ACKNOWLEDGMENTS
Iamindebted to Moira Griffiths for her excellent technical assistance. I would also like to thank Bill Folk and Don Robberson for their helpful discussions and advice, and Beverly Griffin and Elsebet Lund for their valiant effort in helping in the preparation of this manuscript.
LITERATURE CITED
1. Blackstein, M. E., C. P. Stanners, and A. J. Farmilo. 1969. Heterogeneity of polyoma virus DNA: isolation and characterization of non-infectious small super-coiled molecules. J. Mol. Biol. 42:301-313.
2. Cuzin, F., M. Vogt, M. Dieckmann, and P. Berg. 1970. Induction of virus multiplication in 3T3 cells trans-formed byathermosensitive mutant of polyoma virus. II. Formationofoligomeric polyoma DNA molecules. J. Mol. Biol. 47:317-333.
3. Fried, M. 1965. Isolation oftemperature-sensitive mu-tants ofpolyoma virus. Virology 25:669-671. 4. Fried,M. 1970.Characterization of a
temperature-sensi-tivemutant ofpolyoma virus. Virology 40:605-617. 5. Hirt, B. 1967. Selective extraction of polyoma DNA from
infected mouse cell cultures. J. Mol. Biol. 26:365-369. 6. Radloff, R., W. Bauer, and J. Vinograd. 1967. A dye-buoyant density methodforthe detectionand isolation ofclosed circularDNA inHeLacells. Proc. Nat. Acad. Sci. U.S.A. 57:1514-1521.
7. Tai,H., C.A.Smith, P. A. Sharp, and J. Vinograd. 1972.
Sequenceheterogeneity in closedsimian virus 40 de-oxyribonucleic acid. J. Virol. 9:317-325.
8. Thorne, H.V. 1968.Detectionof sizeheterogeneityin the supercoiled fraction of polyoma virus DNA. J. Mol. Biol. 35:215-226.
9. Thorne, H. V.,J.Evans, andD.Warden. 1968.Detection ofbiologically defective moleculesin component I of polyomavirusDNA. Nature(London) 219:728-730. 10. Vogt, M. 1970.Inductionofvirusmultiplication in3T3
cells transformed by a thermosensitive mutant of polyoma virus. I. Isolation and characterization of TS-A-3T3 cells. J. Mol. Biol. 47:307-316.
11. Yoshiike, K. 1968. Studies on DNA from low-density particles of SV40. I. Heterogeneous defective virions produced by successive undiluted passages. Virology
34:391-401.
12. Yoshiike, K. 1968. Studies on DNA from low-density
particles ofSV40. II. Noninfectious virionsassociated withalarge-plaquevariant.Virology34:402-409. 13. Yoshiike, K., and A. Furudo. 1969.HeterogeneousDNA
of simian virus 40. Fed. Proc. 28:1899-1903.
on November 10, 2019 by guest
http://jvi.asm.org/