0022-538X/81/090746-12$02.00/0
Physical Mapping
of
Drug
Resistance Mutations
Defines
an
Active Center of the
Herpes
Simplex
Virus DNA
Polymerase
Enzyme
KARL W.
KNOPF,"12
ELLIOT R. KAUFMAN,3AND CLYDE CRUMPACKER'4*Medical ResearchCouncilVirology Unit,InstituteofVirology,University ofGlasgow,Glasgow,Scotland';
InstitutfurVirusforschung, DeutschesKrebsforschungzentrum,Heidelberg, FederalRepublicofGermany2 Division of HumanGenetics, Children's Hospital Medical Center, and DepartmentofMicrobiologyand MolecularGenetics,Harvard MedicalSchool, Boston,Massachusetts022153;and Divisionof Infectious
Diseases,DepartmentofMedicine,Beth IsraelHospital, and Harvard MedicalSchool,Boston, Massachusetts 022154*
Received21January 1981/Accepted20May1981
The genome structuresofherpes
simplex
virustype 1(HSV-1)/HSV-2
inter-typic recombinants have been
previously
determinedby
restriction endonucleaseanalysis, and these recombinants and theirparental strains have been employed
todemonstrate that mutations within the HSV DNA
polymerase
locus induceanalteredHSVDNA
polymerase activity,
exhibiting
resistance tothree inhibitorsofDNApolymerase. The viral DNA
polymerases
inducedby
tworecombinantsand theirparental strainswerepurified andshown topossess similar molecular
weights (142,000 to 144,000) and similar
sensitivity
tocompounds
whichdistin-guish viral and cellular DNA
polymerases.
The HSV DNApolymerases
inducedby the resistant recombinant and the resistant
parental
strainwereresistanttoinhibition by phosphonoacetic acid, acycloguanosinetriphosphate, and the
2',3'-dideoxynucleoside
triphosphates.
The resistant recombinant(R6-34)
inducedasmuch
acycloguanosine
triphosphate
asdid thesensitive recombinant(R6-26),
butviralDNAsynthesis in infected cells and the viral DNA
polymerase activity
werenotinhibited. The
2',3'-dideoxynucleoside-triphosphates
wereeffectivecompeti-tive inhibitors for theHSVDNApolymerase, and the
Ki
values for thefour2',3'-dideoxynucleoside
triphosphatesweredetermined for the four viral DNApolym-erases. The polymerases of the resistant recombinant and the resistantparent
possessedamuchhigher
Ki
for the2',3'-dideoxynucleoside triphosphates
andforphosphonoacetic acidthandid the sensitivestrains.A
1.3-kilobase-pair
region ofHSV-1DNAwithintheHSVDNApolymerase locuscontainedmutations which
conferred resistance to three DNApolymerase inhibitors. This region ofDNA
sequences encoded foran amino acid sequence of42,000 molecular weight and
definedanactivecenterof theHSVDNApolymerase enzyme.
Thegrowth of herpes simplex virus (HSV) in
the presence of antiviral agents such as
phos-phonoacetic acid (PAA) or acycloguanosine
(ACG) leads to the selection ofdrug-resistant
mutants (13, 25). This resistance is genetically
stableandprovides adistinct phenotypeinHSV
designated PAArandACGr. The mutations
con-ferringresistanceprovidetwo markersforviral
gene functions and the physical locations of
thesemutations,paarand aCgr, have been
de-finedinthe herpes genome (5, 6, 8). The
selec-tion ofHSVtype 1 (HSV-1) andHSV-2 mutant
viruses which are resistant toPAA readily
oc-cursafterpassage of the virus inthepresence of thedrug, is the result of a single mutation (14),
and isdueto analteredHSV DNA polymerase
activity
(5, 13). The guanosine analog ACG[9-(2-hydroxyethoxymethyl)guanine;
acyclovir]re-quiresphosphorylation toexertits antiviral
ef-fect (9, 11). The virus-coded deoxypyrimidine
kinaseproduces phosphorylationtoACG
mono-phosphate, and a subsequent conversion to
di-andtriphosphatespresumably occursbycellular
pathways (11). The ACG triphosphate
(ACG-TP) is a main metabolite ofinfectedcellsand is
an effective inhibitor ofwild-type (WT) HSV
DNApolymerase(11,12, 26).GrowthofHSV in
the presence of ACG selects for resistant mu-tants; thisresistanceiscontrolled bytwo genetic loci in the HSVgenome, the viral
deoxypyrimi-746
on November 10, 2019 by guest
http://jvi.asm.org/
dinekinaseand theviralDNApolymerase loci
(7,25).
Restriction endonuclease analysis of the
ge-nomes of plaque-purified HSV-1/HSV-2
inter-typicrecombinants has permitted the physical
map limits for two distinct regions controlling
ACGtobedefinedwithin the HSVDNA
polym-eraselocus (8).One of these regions contains the
mutation for acgrclosely linkedtothe mutation
forpaar, and these mutationsarefound within
a1.3-kilobase-pair region of HSV DNA for
HSV-2and withina2.6-kilobase-pair region for
HSV-1. The other region is found within adjacent
sequences of HSV DNA and defines the
type-specific sensitivity of HSV-1 and HSV-2 ACG.
These regionsarefound inequivalent positions
within the HSV-1 and HSV-2 genome in the
areadefinedasthe HSVDNApolymerase locus.
Twoof theseintertypic recombinants contain an
HSV-1 DNA insertion in an otherwise HSV-2
genomeanddiffer significantly intheir
sensitiv-itytoACGandPAA.Theserecombinants were
derived by markerrescue of
temperature-sensi-tive (ts) mutations and resulted from a cross
between the DNA of HSV-2 ts6 (HG-52) and
HSV-1 strain 17 (ts+, paar_1) (R6 series) (5).
The R6-26 recombinant is sensitive to plaque
inhibitionbyPAA andACG,whereas the R6-34
recombinant exhibits PAA' and ACGr. In this
report the viral DNA polymerases induced by
these recombinants and their parental strains
have been partially purified and tested for
sensitivity and resistance to ACG-TP, PAA,
and the known chain-terminating compounds,
the
2',3'-dideoxynucleoside triphosphates
(didNTP's). This report correlates genome
structuresand thepresenceof thegenemarkers
for
paar
andacgr
with the function ofpurifiedHSVDNApolymerase in thepresenceofthree
compounds of dissimilar structure. A
1.3-kilo-base-pairregion of HSVDNA within the viral
DNApolymerase locuscontains a mutation or
mutationsconferring resistance to three DNA
polymerase inhibitors. The DNA sequences
within thisregion encode foranactivecenterof
theherpes
simplex
virus DNApolymerase.(This workwaspresented inpart atthe
Inter-national ConferenceonHerpesViruses,Atlanta,
Ga.,
on 21 March 1980 and at the 5th ColdSpring Harbor Workshop on Herpes Viruses,
Cold
Spring
Harbor, N.Y.,on31August1980.)MATERIALS AND METHODS Materials.Acyclovirwas agenerousgift fromJohn
Beale of Burroughs Wellcome Ltd., Beckenham, UnitedKingdom.PAA was agift from LaceyOverby of AbbottLaboratories, Inc., NorthChicago, Ill. Acti-vated salmon spermDNAprepared by mildpancreatic
DNase digestion as previously described (29) was a
kind gift of Helen Moss-Rixon, MRC Virlogy Unit, Glasgow. Deoxynucleoside triphosphates (dNTP's), didNTP's, and oligodeoxyguanidylic acid-polydeoxy-cytidylic acid [oligo(dG)1218-poly(dC)] were from P.
L. Biochemicals. Radiochemicals were from Radi-ochemicalCenter, Amersham.[3H]ACG was prepared by NewEngland Nuclear Corp.
Cells and viruses. Baby hamster kidney (BHK 21-C13) and Verocells were grown in Eagle medium supplemented with 10% tryptose broth and 10% bovine calf serum (21). Cell monolayers (107cells) in 90-mm plastic petri dishes (Falcon Plastics) were used throughout. Virusinfection was carried out with 10
PFU/cellinmedium without tryptose. All virus strains usedwerederived from stocksprepared inGlasgow by low-multiplicity passage in BHK 21-C13 cells. The temperature-sensitive mutant of HSV-2, ts6 (strain HG-52), has been previouslydescribed(27).The PAA' mutantofHSV-1 strain17 (ts+,paar_j) was isolated by passage in the presence of PAA (100mg/ml) and
wascharacterizedpreviously (13). The HSV-1/HSV-2 intertypic recombinants R6-26 and R6-34werederived by crossesbetween HSV-2 ts6 (strain HG-52) DNA and restrictionendonuclease-cleaved DNA from
HSV-1 strain 17 (ts+,paa'r1) DNA, isolated by marker rescue,andplaque purified at38.5°C (6).
Preparation of cell extracts andpurification
ofHSV DNA polymerase. All infected cells were
harvestedat 18 hpostinfectiontopreparecytoplasmic
extracts. To demonstrate that the resistant
recombi-nantR6-34 induces DNApolymerase activity which is resistanttoACG-mediated inhibition, cytoplasmic ex-tracts were prepared from confluent baby hamster kidney cells (BHK-C13) infected with WT HSV-2 (strainHG-52),R6-26,orR6-34(multiplicityof infec-tion, 10 PFU/cell), and at 7 h postinfection 20 uM ACG inwater wasaddedtothe infectedcells,orwater
alonewasaddedtocontrol dishes. The infected cells
wereincubated in the presenceorabsence of ACGat
37°C,andcultureswereharvestedat 18h postinfec-tion.Cellextracts wereprepared bylysingcells in0.25
M potassiumphosphate buffer (pH7.5) containing1
mM each ofdithiothreitol, EDTA, ethylene glycol-bis(B-aminoethyl ether)-N,N-tetraacetic acid, and 0.5% Triton X-100. After centrifugation to remove
nuclei (15,000xg for1hat4°C), aliquotsofcytosol extracts werekept in35% ethylene glycol at -20°C
until use. These extracts were employed directly to
assayHSV-induced DNApolymerase activityby
mea-suring theincorporation of[32P]dGTP into the syn-thetic DNAtemplateprimer,oligo(dG)1218.poly(dC).
Forpurification of HSV DNA polymerase, 200-1i samplesoffresh cellextracts(600 ,ugofprotein)were
layeredonlinearglycerol densitygradients (20to50% glycerol in 0.25 M potassium phosphate buffer [pH
7.5] containing 1 mM dithiothreitol and 0.1 mM
EDTA), velocitysedimentationwascarriedout ina
Sorvall TV885rotor(45,000xg for15hat2°C)with appropriate markers for molecular weight, 0.25-mi fractionswerecollected,and10,ulof eachfractionwas
assayedforDNApolymeraseactivity (17).
DNA synthesis.Infected cellsweregrown in the
absenceorpresence of5jAMACGin 10mlof medium
on November 10, 2019 by guest
http://jvi.asm.org/
748 KNOPF, KAUFMAN, AND CRUMPACKER
containing 87.5,iCi of carrier-free "Pi. Cells were harvested 18 h postinfection by scraping them into coldreticulocyte standard buffer. After being pelleted, cellswere suspended inreticulocyte standardbuffer containing 10 mMEDTA, 1% sodium dodecyl sulfate, and0.5mgofproteinase K per ml and incubatedat
500Cfor3h. Then DNAwasphenol extracted twice andprecipitated with alcoholat-20°Covernight.The washedand dried DNA pelletsweresuspended in 10 mM Tris-hydrochloride (pH 8)-10 mM EDTA con-taining 100,ugof RNaseAper mland 1,000 U ofTi RNase (Worthington Diagnostics) per ml and incu-bated for3 h at370C. The DNAwasalcohol precipi-tated, washed, and dissolved in 50 mM Tris-hydro-chloride (pH 8)-10 mM EDTA, and CsCl was added to give a final density of 1.715 g/cm3. Equilibrium density CsCl gradient centrifugation was carried out in aSorvall TV865 rotor at 43,000 rpmfor 18 h at 200C. Gradientswere dripped from the bottom, and fractionswere collected directly ontoWhatman 3M paperslips.Determinationof trichloroacetic acid-pre-cipitable radioactivitywasperformedby the Bollum technique (4).
Measurement of ACG-TP. To determine the
amountofACG-TP formed in infectedcells,[3H]ACG
(50,tCi/ml; 200,Ci/mmol; New England Nuclear
Corp.) was added at 7 h after infection, cells were incubated at370C until 18hpostinfection and then harvested, andextracts wereprepared. Medium was removed, the disheswerewashedthree times in0.5ml ofice-cold 0.85%NaCl,andsoluble nucleotideswere
extracted in 0.5 ml of 0.4 MHCl04for30min at40C. Theextracts wereneutralized with KOH and concen-tratedbylyophilization. The ACG-TP was separated
onpolyethyleneiminecellulosethin-layer chromatog-raphy sheets developed with 2.0 M LiCl-2.0 N HCOOH (1:1, vol/vol). The chromatography sheets
were fractionated, and the triphosphate was eluted from the adsorbent with1ml of0.1NHCland counted ina liquid scintillation counter. The protein in the cellular extracts was determined by the method of Lowry et al. (20), and the results are expressed as picomoles of nucleotide formed per microgram of cell protein.
The[3H]ACG wasobtained by tritiation of ACG, and then [3H]ACGwas purified by ascending chro-matography onWhatman 3M paper in
N-propanol-water(7:3).TheRf for ACG in this system is 0.53 (11); this corresponded to the onlyUV-absorbingmaterial observed on the chromatogram. The [3H]ACG was
eluted andrechromatographed before use.
HSV DNA polymerase assay. When the syn-thetic template primer,
oligo(dG)12118poly(dC),
was employed, the standard reaction mixture contained (inafinal volume of 100,l): 50 mM Tris-hydrochloride (pH 8.0), 50,ug of bovine serum albumin, 0.5 mM dithiothreitol, 7.5 mM MgCl2, 100 mM ammonium sulfate,0.01mM[32P]dGTP(1,450cpm/pmol), and 50
jig
ofoligo(dG)1218.poly(dC) per ml. With activated DNA as the template primer, thestandard reaction mixture contained (in a final volume of 100pl):
50 mM Tris-hydrochloride (pH 8.0), 50jig
of bovine serum albumin,0.5mMdATP, dCTP, dGTP, and[32P]dTTP (200 cpm/pmol), and 100itg
ofactivated salmonsperm DNA. Under these conditions, all DNA polymeraseassaysexhibited linear kinetics of incorporation for 20 min.Incubationwasperformed at370C for the indi-catedtime, andsamples of the reaction mixturewere
spottedontoWhatmanGF/C glass ifiters of Whatman
no. 1filter paper,precipitated by 10% trichloroacetic acid, and treatedtodetermine acid-insoluble radioac-tivity as previously described (17, 24). One unit of HSV DNA polymerase activity is defined as the
amountof enzymecatalyzingthepolymerizationof 1 nmol ofdeoxynucleotides per h under standard assay conditions.
Alkaline DNase assay. Alkaline DNasewas
mon-itored as described by Morrison and Deir (23) by following the release of acid-soluble products from [3H]thymidine-labeled DNA. The reaction mixture contained(inafinal volume100tl):50M Tris-hydro-chloride (pH9), 5 mM MgCl2, 10 nM dithiothreitol, and 10 ,ug ofdouble-stranded, [3H]thymidine-labeled BHK21-C13 DNA (6,250cpm/,ug)and either5-, 10-,
or
20-pl
samples of cellextractin 200A1
ofasuspension ofHyflo-SuperCel (20 mg/ml;Koch-LightLabs)in 1 Mperchloric acid. Aftermixingandcentrifugationat10,000xgfor2.5min,150-,lI samplesof the superna-tants werecounted in10mlofAquasolcocktail. Under thegivenconditions, 1 U of alkaline DNaseactivityis definedastherelease of1 nmol ofdeoxynucleotides per hat37°C.
Thymidine kinase assay. Thymidine kinase activity was assayed by measuring the amount of [3H]thymidine phosphorylated by an extract of in-fected cellsby the method of Jamiesonetal.(15, 16). Briefly, the reaction mixture contained (in a final volume of100tl): 20mM potassium phosphate (pH 7.5), 10mM MgCl2, 10,LM [3H]thymidine (10 ,uCi/
!Lmol),
and cellextract ataconcentration of 50 to 100,ug ofproteinperml.After incubation for 30minat
370C, radioactivity bound to Whatman DE81 filter disks wasdetermined. One unit ofthymidine kinase activity is defined as the conversion of 1 nmol of thymidine intoaphosphorylatedform within 1 hat
370Cunder the stated assayconditions. Protein
con-centrationsweredeterminedbythe methodofLowry
etal. (20).
RESULTS
Development of HSV-1/HSV-2 intertypic recombinants and determination of the
HSV DNA
polymerase
locus.Seventy-four
HSV-1/HSV-2 intertypic recombinants have
been generated by marker rescue of
tempera-ture-sensitivemutantsexhibiting defectiveDNA
synthesisat thenonpermissive temperature (5,
6).The genome structuresoftheserecombinants
havebeen determinedby restriction
endonucle-aseanalysis ofthegenomes after plaque
purifi-cation andhavebeen employed in determining
thephysicalmap limits of five
temperature-sen-sitive mutations (ts) and mutations conferring
resistance to PAA (paar) and ACG (acgr) in
both HSV-1 and HSV-2 (5, 6, 8). The genome
structuresfor the recombinants resulting from
crossesbetween HSV-2ts6 (HG-52) and HSV-1
strain 17(ts+, paar_j) (R6series)wereanalyzed
on November 10, 2019 by guest
http://jvi.asm.org/
withsevenrestrictionendonucleases in the
re-gion from30 to 50 map units,enabling the
phys-icalmaplimitsof the HSV-2 ts6mutation and
thepaa-1 andacg-1 mutations of HSV-1 (strain
17) to be established (6, 8). Two of these R6 recombinants, R6-26 and R6-34, were selected
for further analysisto determine whether
mu-tationswithin theHSV DNA polymerase locus
which confer resistance to plaqueinhibition by
ACG and PAA induce an altered HSV DNA
polymerase activity. These two recombinants
wereselectedbecause they possesssimilarsmall
HSV-1 DNA insertions inan otherwise HSV-2
genome.ThisHSV-1DNA correctsthe ts6
mu-tationpermitting bothrecombinants toreplicate
atthenonpermissivetemperature, but these two
recombinants differ widely in their sensitivity to
PAAand ACG. The R6-34recombinant has an
HSV-1 DNA insertion extending fromthe
HSV-1 BglII i-d site to the HSV-1 EcoRI m-o site
(mapunits39.6 to 42.8, including the region of
uncertainty), is PAAr (efficiency of plaqueing,
0.6inthepresence and absence of 100lg of PAA
per ml) and markedly ACG' (50% inhibitory
dose, 17.5
,tM).
The R6-26 recombinant, on theother hand, contains HSV-1 DNA sequences
extending fromtheHSV-1 BgllI i-d site to the
HSV-2 Bam h'-j' site (map units 39.6 to 41.0,
including theregion of uncertainty), but is still
PAA8 (efficiency of plaqueing,0.4 x 10-4in the
presenceand absence of100 ,ugofPAA per ml)
and ACG8 (50% inhibitory dose, 0.12 ,uM) (8).
The genome structures of these two recombi-nants and their restriction endonuclease sites
have been previously published (6, 8). The
re-combinants and their parental strainswere
em-ployed in all infections, and their polymerases
were
partially
purified and compared.Effect
of
ACGonviral and cellular DNAsynthesis
ininfected cells. The recombinantandparental strains differed markedly intheir
sensitivityto PAAandACG when analyzed in
a plaque reduction assay. This efficiency of
plaqueing in the presence of5 ,uM ACG
com-pared with that in the absence of inhibitorwas
82%forR6-34, <0.5% for
R6-26,
45% forPAAr_1,
and 9% for HSV-2 ts6 (8). Since the effect of
PAA on DNA
synthesis
ofparental
strainsandPAArmutants has been
previously
determined(13),it was of interest toalsomeasurethe effects
ofACGonviral and cellular DNAsynthesisin
cells infected with the recombinants and their
parental strains. To show that the presence of
the
acgr-1
mutationaffectstheability
of infectedcellstosynthesizecellular and viral DNA in the
presence of ACG, the DNA
profiles
fromin-fectedcells in the presence and absenceof ACG
were compared. Confluent cells (BHK) were
infected in the absence orpresence ofACG
(5
,uM), and theincorporation of 32p, into DNAwas
determinedat 18hpostinfection by analysis of
total cellular DNA on CsCl density gradients
(Fig. 1). In thepresenceof the drug, viralDNA
synthesisof the ts6 parent and the R6-26
recom-binant washardly detectable. Viral DNA
syn-thesis of the
PAAr_1
parent andthe R6-34re-combinantwere resistant toACG, with20 and
23%,respectively, of viralDNAbeingpresentin
the absenceof thedrug. CellularDNAsynthesis
wasdecreased byACG in all four infections, but
40% ofcellularDNAsynthesis remainedin the
presenceof5
,uM
ACG. For both the PAAr-1 andthe R6-34 recombinant,viral DNAsynthesisis
inhibited to a greater extent by ACG (5 ,uM)
than is plaque formation,
indicating
that theACGr virus may be more effective in utilizing
viral DNA in the formation of infectious virus.
Little of the newly synthesized viral DNA
ap-pears toberequired for effective packaging into
infectious virus.Adirectcomparison of the
effi-ciency ofplaqueing and viral DNA
synthesis
inthepresence ofACG isnot
possible
fromthesetwoexperiments because the cellswereinfected
atdifferentmultiplicities and
analyzed
atdiffer-
5--S
x
E
z
05
a-C'l4
(Y)
HSV
A I
BHK
f\uj
HSV BHK
B I I
I~~~~~~~~~~~~~~~~~~~~~~~~~~~~
20 20
IC
1 5 0D I I_v\S~~~~~~~I
10 2-0 3-0 10
Fraction number
20 30
FIG. 1. Equilibrium density gradients of DNA from infected cells in the presence and absence of
ACG. BHK cellswereinfectedwith HSV-2
(HG-52)
ts6(A), HSV-1 PAAr-l (B),R6-26(C), orR6-34(D)
virusandgrowninthepresence(0)orabsence(0) of5 MACG and32p;.At18hpostinfectioncellswere
harvested, and DNAwasextracted andanalyzed by
CsCIdensitygradientcentrifugation.Fractionswere
collected andprecipitatedin 5% trichloroaceticacid,
andradioactivitywasdetermined. The DNAprofiles
obtained in thepresence and absence
of
ACG areplotted on the samefigureforcomparison. Arrows indicate thedensityofHSVDNA (1.729
g/cm3)
orcellular DNA(1.705
gm/cm3).
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.499.255.449.335.525.2]enttimes after viral infection.To radiolabelwith 32P, the cellshad to be grown in P04-free
me-dium aswell. However, the R6-34recombinant
and thePAAr-1 parentalvirusclearlyshowthe highest resistance of viral DNA synthesis and
thehighest efficiency ofplaqueing inthe
pres-enceofACG.
An altered viralDNApolymeraseconfers resistancetoACG-mediatedinhibition.The extractsfrom HSV-2 WT(HG-52)-, R6-26-,and R6-34-infected cells incubated in the presence
and absence of ACG (20
,tM)
wereprepared at18h postinfectionwhen ACG had beenpresent (7 to 18 h). These cytoplasmic extracts were
employedas an enzyme source in the assay of
HSV DNA polymerase activity by using
oligo(dG)1218s*poly(dC)
as atemplate primerandmeasuringtheincorporation of[32P]dGTPinto
acid-insoluble material after 10 min at 370C. With each of thecellular extractsobtained with-outACG treatment, thekinetics of polymeriza-tion proceeded linearly for 20 min. When
in-creasing amounts of cellular extract were
em-ployed in the DNA polymerase assay alinear
increase inincorporation of[32P]dGTPintothe synthetic template was also observed; this in-crease was almostcompletely inhibitedby
ex-tractsprepared from ACG-treatedHSV-2
WT-or R6-26-infected cells (Fig. 2A and B). The extract prepared from ACG-treated, R6-34-in-fectedcells,however,exhibitedlinearkineticsof DNApolymeraseactivitysimilartothoseofthe extract of R6-34-infected cellsprepared in the absence of ACG (Fig. 2C).
To exclude the possibility that the ACG-re-sistant DNApolymeraseactivity inducedby R6-34 was merely due to the failure of
R6-34-in-fected cells to make an active inhibitor, the
amountofACG-TP formedincellsinfected with
R6-26 and R6-34 in the presence of[3H]ACG
wasmeasureddirectlybypolyethyleneimine
cel-lulose chromatography. It has been previously shownthat theACG-TPis the activeinhibitor
of HSV DNA polymerase (11, 12, 26). The
amount ofACG-TP formedin 26- and R6-34-infected cellspermicrogram ofcellular
pro-teinwasverysimilar andwasmuchgreaterthan
thatformedinuninfected Verocells(Table1).
To confirm that the R6-34-infected cells
in-duce an active inhibitor of DNA polymerase
activity,the ability ofheat-inactivated extracts
fromACG-treated,infectedcellstoinhibit HSV
DNApolymerase wascompared. When 10,ulof
aninfected cell extractwas heatedat600C for
10min and then addedtoanactivecellextract,
little inhibition was observed (97.9% of DNA
polymeraseactivity remaining).Anextract,
pre-pared inthepresence of ACG (20,uM,7to18h
5 10 15
Cell extract(,ugprotein)
FIG. 2. HSV DNApolymeraseactivityofextracts
ofBftKcellsinfectedwithrecombinant virusesinthe
presence and absence of ACG. Cells were infected
withHSV-2(HG-52) WT(A),R6-26 (B), orR6-34(C)
in theabsence (0) orpresence(0) of ACG(20IpM), and extractsprepared at 18 hpostinfection. HSV DNApolymerase activitywasdeterminedby
measur-ing[32P]dGMPincorporatedwithanoligo(dG),1218
poly(dC) template primer in standard assayfor 10
minat370CasdescribedinTable3.Protein
concen-tration was determined by method ofLowryetal.
[image:5.499.314.408.65.280.2](20).
TABLE 1. FormationofACG-TP in cellsinfected
withrecombinantvirusesa
ACG-TP TotalACG-TP formed
Virus
formed(pmol)(pmol/ug
of cellprotein)R6-26 100.5 5.0
R6-34 81.0 4.9
Vero alone 2.0 0.14
aConfluent Vero cells in 90-mm dishes were in-fected at amultiplicity of infection 10, and at 6 h [3H]ACGwasadded(200,uCi/mmol).Cells were har-vestedat 18h andextracted withHC104, and ACG-TP was determined by polyethyleneimine cellulose chromatography. Protein in cellular extracts deter-minedbymethod ofLowryetal.(20).
postinfection) andinactivated, produced 59.9%
inhibition ofthe DNA polymerase activity
in-duced by HSV-2 WT-infected cells. This same
levelofinhibition wasobservedafter the
addi-tion ofinactivatedextractspreparedfrom
ACG-treated, R6-26-orR6-34-infectedcells (Table
2).
Thus, infectionwith theWT HSV-2orthetwo
recombinants containing a single HSV-1 DNA
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
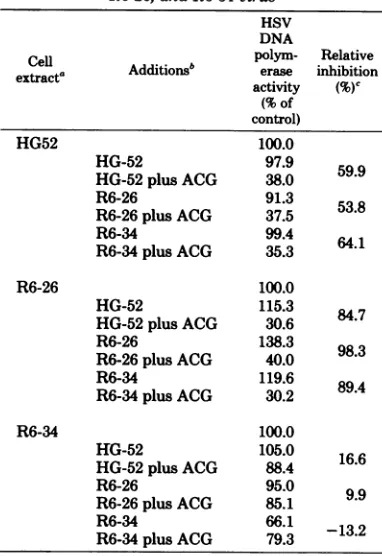
[image:5.499.265.452.424.494.2]TABLE 2. Effect of ACG on HSV DNA polymerase activityofextractsofBHK cellsinfectedwithHG52,
R6-26, and R6-34virus
HSV DNA
Cell Additionsb polym-erase inhibitionRelative
extracta activity (%)C
(% of control)
HG52 100.0
HG-52 97.9 59.9
HG-52 plusACG 38.0
R6-26 91.3
R6-26plus ACG 37.5 53.8
R6-34 99.4
R6-34plus ACG 35.3 64.1
R6-26 100.0
HG-52 115.3 84.7
HG-52plusACG 30.6
R6-26 138.3 98.3
R6-26plus ACG 40.0
R6-34 119.6
R6-34plus ACG 30.2 89.4
R6-34 100.0
HG-52 105.0 16.6
HG-52plus ACG 88.4
R6-26 95.0 9.9
R6-26plus ACG 85.1
R6-34 66.1
R6-34plus ACG 793 -132
aBHK
cells
wereinfected with the indicated virus and grown in the absenceorpresence ofACG(20,M) from 7 hpostinfection, andcellextract waspreparedat 18 h postinfection. Heat-inactivated extract was
prepared by heatingat
600C
for30mintoinactivate DNApolymerase.Samples(10pd)
ofextractaloneorwith10
pd
of inactivatedextract wereincubated for 10 minat370C
under HSV DNApolymeraseassaycon-ditionsusing50
Mg
ofoligo(dG)12-18-poly(dC) astem-plateand0.01mM
['P]dGTP
(1,450cpm/pmol).The values for 100% represent theincorporation of71.4, 23.5, and12.1pmolof dGMP per,g ofcellproteinfor HG52;R6-26; and R6-34-infectedcells, respectively.b InactivatedcellextractandACG-treated cell ex-tract wereaddedasindicated.
eRelative inhibitionbyACG-treated inactivated ex-tractsisthedifference obtained inpolymeraseactivity ofagivenextractassayedwithmock-treatedor ACG-treated inactivatedcellextracts.
insertion in an otherwise HSV-2 genome induced
similar amounts ofan active inhibitor of HSV
DNA polymerase
activity.
The DNApolymer-aseactivity induced
by
the R6-26recombinantwas extremely sensitive to inhibition
by
theheat-activated extracts,
revealing
an 84to 98%decrease in DNA
polymerase
activity.
The DNApolymerase
activity
of the HSV-2 ts6 mutantwasinhibitedby58.9%
by
inactivated extracts,afigure verysimilarto the inhibitionobserved
for HSV-2 WT DNA
polymerase.
Extractsfrominfectedcells alone
produced
noinhibition.The DNA
polymerase
activity
inducedby
theACG-resistant recombinant R6-34or the PAAr_
1HSV-1parentviruswas
markedly
resistanttoinhibition byinactivatedextracts fromcells
in-fectedinthepresence of ACG. In the absence of
anyinhibitor, 12.1pmolof[32P]dGTPper
lag
ofproteinwas incorporated intoDNA
by
theex-tract from R6-34 infected cells; this was de-creased by only 16.6 to 9.9% after the addition of extractsfrom ACG-treated HSV-2 WT-, or R6-26-infected cells (Table 2). In the presence of an
inactivatedextractfrom ACG-treated,
R6-34-in-fected cells,the R6-34-induced DNApolymerase
was actually stimulated by 13%. The PAA-r
HSV-1 parent DNA polymerase wasinhibited
by only 11.7%.Thus, the R6-34 recombinant and
its
PAAr_1
parent induced HSV DNApolymer-ase activity which was resistant to the
ACG-induced inhibitorpresentin infected cell
cyto-plasma.This cytoplasmic inhibitor is ACG-TP,
and the resistance of the R6-34-induced DNA
polymerasetoACG-TPis due to an altered viral
DNA polymerase.
The
altered
viralDNApolymeraseisre-sistanttoinhibition by didNTP's.The HSV
DNA polymerasewas purified from BHK
cells
infected withthe recombinants and their
paren-tal strainsby
sedimentation
in glyceroldensitygradients (Fig.3).Allfoursedimentation
exper-iments revealed a major peak of DNA
polym-erase activity sedimenting only slightly slower
than the
sedimentation
standard,glyceralde-hyde-3-phosphate dehydrogenase (molecular
weight, 144,000). The peak has a molecular
weightofapproximately 142,000 to 144,000, the
estimated molecular weight of the HSV DNA
polymerase (17, 24). The major alkalineDNase
activity and the dNTP'swereclearly separated.
Thepeak fractions possessingthe DNA
polym-erase activity were further examined for their properties in high salt and with inhibitors of
DNA polymerase activity, N-ethylmaleimide
and PAA, two inhibitors previously shown to
provide sensitive criteria for discriminating HSV
DNA
polymerases
fonn cellulara and ,B DNApolymerases (28). All four of theDNA
polym-eraseswere
clearly
stimulatedin the presenceof100
MuM
ammoniumsulfateby5- to10-fold whenactivatedsalmonsperm DNAwas
employed
asa
template
(Table
3).Aconcentration of2.5,uM
N-ethylmaleimide
inhibited the observed DNApolymeraseactivityobtained fromallfour
infec-tions by greater than 95%
(Table 3). By
the criteriaof molecularweight,
stimulationby
high
salt,and inhibition
by
N-ethylmaleimide,
there-fore,theobservedDNA
polymerase activity
on November 10, 2019 by guest
http://jvi.asm.org/
752 KNOPF, KAUFMAN, AND CRUMPACKER
1 2 3 4
5A
I D
[image:7.499.62.252.62.252.2]10 20 30 10 20 30 Fraction number
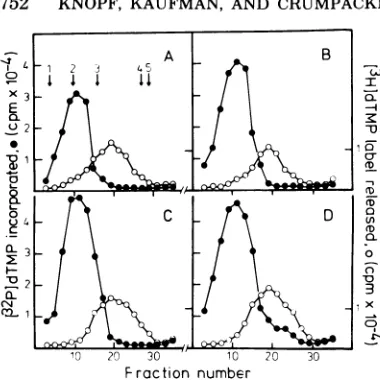
FIG. 3. Purification ofHSVDNApolymerasefrom parental and recombinant virus-infected cells.
Ex-tractsfrom cellsinfectedwith HSV-2(HG52)ts6(A), HSV-1 PAA'-I (B), R6-26(C), and R6-34 (D) were
layeredonlinearglycerol gradients(20to509%)and centrifuged (Sorvall TV885rotor, 45,000 rpm,2°C,
15h). Fractionswerecollected,and 10-,ilsamplesof eachfractionwereassayedforHSVDNApolymerase with activated salmon sperm DNA as a template (-).Alkaline DNaseactivity(0)wasalso determined under standard incubation conditions in10-Ml
sam-ples ofeach fraction. Sedimentation markers were
included in eachgradientandareindicatedas fol-lows: 1,catalase; 2,glyceraldehyde-3-phosphate de-hydrogenase; 3, bovineserumalbumin; 4, cytochrome c;and 5,amixtureof[32P]dATPand[32P]dGTP.
sessed the properties of the HSV DNA
polym-erase enzyme,andthe DNApolymerase
activity
obtained after all four infections
appeared
tohave asimilar molecularweight.
The structureofACG-TP is similartothat of
the didNTP's in that these nucleoside
analogs
lacka3'-OHgroup,preventingchainelongation
(2). The didNTP's are well-established
inhibi-tors ofDNA
synthesis
which becomeincorpo-rated intotheprimer andproduce chain
termi-nation. The four didNTP's were compared for
theabilitytoinhibit the viralDNA
polymerase
from the two recombinants and their
parental
strains. Initial experiments
employing
oligo-(dG)12
18-poly(dC)
as atemplate primer
tomea-sure the
incorporation
of[32P]dGTP
indicatedthat theR6-34-inducedDNA
polymerase
incy-tosol extracts was resistant to inhibition by
didGTP. ViralDNApolymerase activity in
ex-tracts of HSV-2 WT (strain
HG-52)-,
R6-26-,andR6-34-infected cellswascompared, and the
concentration of didGTPrequiredtoinhibit 50%
of theDNApolymerase activity (50% inhibitory
dose) wasdetermined.The 50%inhibitory dose
values forthe
viral
DNApolymerase induced byHSV-2, R6-26, andR6-34 were5, 1, and 74,uM
ofdidGTP,
respectively (data
notshown).
Thus,
theR6-34DNApolymerase activity requirad74
timesmoredidGTP for 50% inhibition
tha_lid
theDNA
polymerase
inducedby R6-26.The inhibition constants
(Ki)
for all fourdidNTP's with activatedDNA weredetermined
with the
purified
viral DNA polymerase fromTABLE 3. Template characteristics and apparent kineticconstantsofthepartiallypurifiedHSV DNA polymeraseenzymesofparental and recombinantviruses
HSV DNApolymerase activity (pmol
in-corporated) under thefollowingcondi- Km(AM) Ki(ILM)b
tionsa:
Virus SS
SSDNA DNA Oligo(d
SS DNA plusAS plusAS G)1218. dNTP's dGTP didNTP's PAA
plus poly(dC)
NEM
ts6 11.2 57.1 1.4 27.5 10.8 22.3+5% 22.6 8.2
PAA'-_1 13.5 128.2 6.1 57.0 11.4 22.3±5% 122.5 37.5
R6-26 9.1 85.2 1.0 25.8 11.9 22.3+5% 14.7 4.5
R6-34 14.6 142.9 2.4 69.9 10.9 22.3+5% 135.1 39.0
a HSVDNApolymeraseactivity was determined under standard incubation conditions (20 min at
370C)
with either 100,ugofactivatedsalmonsperm(SS)DNApermlor 25jg
ofoligo(dG)*poly(dC) per ml with or without 100mMammoniumsulfate (AS) and 2.5 mM N-ethylmalemide (NEM). The source of viral DNA polymerase wasthepeak fraction ofenzyme activity from the glycerol gradient shown in Fig. 3.bTheKmdata fordGTPanddNTP's and theKidata fordidNTP's were derived from Fig. 4 and 5. The amountsof protein usedwere6.2, 6.4, 5.9, and 6.9 gforts6,PAAr_1,R6-26, and R6-34,respectively. For theKm
andKidetermination,one-halftheamount of enzyme was used. TheKifor PAA wascalculated fromgraphsby
plottingPAAconcentrationversus
Vm.,/v
on a Dixon plot. TheKmfor dGTP wasderived from Fig. 5 by using 25jug
ofoligo(dG)1218 poly(dC)permlas a template.4 0
x3
02
0
.1
.84
OL 3
I
2
(L
cn 1
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.499.65.455.445.562.2]HSV
therecombinants and theparentalstr
4 and Table 3). The units of initial
velocity (v) were expressed as pico
dTMP incorporated per microgram c
duringa20-min assay; enzyme
activit3
ear over 20min. Inthe Lineweaver-Bi
ble-reciprocal plot of1/v versus
1/(dN
competitive inhibition-type kinetics
exy
a chain-terminating
compound
were(Fig. 4). All four of the didNTP's had
Ki
for activated DNA. TheKi
for eadidNTP's with viral DNA
polymerase
estfor R6-26
(Ki,
14.5,iM) followed bts6
(Ki,
22.6,M) andwashighest
withfied DNApolymerases from HSV-1, Pi
122.5
,M)
and R6-34(Ki,
135.1,uM).
0.051
-01
0.1
A
a
I
VI
B
0.05-0.1 0.2 -0.1
C
lip-1
V
ains (Fig. The peak fractions ofpurified DNA
polym-I enzyme erase werealso employedto testforthe
sensitiv-imoles of ityof theenzyme activity to inhibition by PAA.
)fprotein Activatedsalmxon spermDNA was employed as y was lin- a template; the reactions were linear over 20
arke dou- min,and the
Ki
for PAA was determined fromiTP), the the Dixon plot of
V./v
versus PAA. TheKi
forpected
for PAA was again lowest for R6-26(Ki,
4.5,iM)
observed followed by HSV-2 ts6
(Ki,
8.2AM)
and wasthesame highest with the purified polymerases from
ch of the HSV-1
PAAr_1
(Ki,
37.5,M) and R6-34(Ki,
39.0!was low- tiM) (Table 3). The comparative
Ki
values foryHSV-2 PAA and for the four didNTP's with the
paren-Lthepuri- tal and recombinant DNA polymerases corre-A,A 1
(Ki,
lated extremelywellwith the resistance of theseenzymes to ACG-TP, to the resistance of viral
DNA synthesis in thepresence of ACG, and to
theresistance of HSV plaqueformationto ACG
(8).
To exclude the possibility that resistance of
viral DNA polymerase to ACG-TP, PAA, and
* didNTP's is due to an altered
affinity
of theresistantpolymerasefor dNTP's, theKmvalues
* for each of the dNTP's were determined by
using
activated salmonsperm
DNA oroligo-0.1
0'2
(dG)1218
poly(dC)asatemplate. TheKmvalueswere
essentially
identical forall four dNTP's orfor dGTP with the synthetic template for all
four viralpolymerases (Table3and Fig. 5). The
altered viralDNApolymerase activity was not
dueto analtered Km.
0.051
0.1 0.2 -01
I
dNTP
]-1
,(
pM)-1
D
1
V
1
0.1 02 FIG. 4. Determination ofKm fordNTP's andKi
for didNTP's of parental and recombinant HSV DNApolymerases. HSV DNA polymerase activity
wasassayedonthepurified peak fractions from Fig.
3 under standard incubation conditions (20min at
37°C) with 100pgofsalmonspermDNApermlby usingallfourdNTP'satequivalentconcentrations. Therateof incorporation of[32PJdTTPin percent in the absence(0) orpresence(0) of50 mMofeachof
thefourdidNTP'swasusedasthe initialvelocity (v)
in thedouble-reciprocalLineweaver-Burkeplot.The
enzymeactivitywaslinearover20min.In the absence
of inhibitor, 100% represents specific activity of 9.2, 18.4, 15.4, and 22.8pmol ofdTMPperpgof protein for ts6 (A), PAArX1 (B), R6-26 (C), and R6-34 (D), respectively.
0.01
I I
-0.04 0.01 0.02 0.03 0.04
(dGTPJ1.
(UM)1
FIG. 5. DeterminationofKmfordGTP. Thepeak fractions from Fig. 3wereemployedtomeasurethe HSV DNApolymerase activityin theparentaland recombinantviruses. HSV DNApolymerasewas
as-sayed understandard conditions (10min at 37°C)
witholigo(dG)1218.poly(dC) (25 pg/ml)asatemplate,
and therateof incorporation of [32P]dGTPin per-centageof complete incorporation wasused to cal-culateinitial velocities inthedouble-reciprocol plot.
The specific activities for the DNApolymerase of
HSV-2ts6(0),HSV-PAAr_l (0),R6-26(A),and R6-34 (J) were 0.23, 0.78, 0.33, and 0.19 pmol/,ug of proteinpermin, respectively.
0 05.
-0.1
0
I
I I I IVOL. 39,1981
0.02r
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.499.49.240.249.502.2] [image:8.499.255.447.396.527.2]754 KNOPF, KAUFMAN, AND CRUMPACKER
To exclude thepossibility that the recombi-nantsdiffered in the expression of viral deoxy-pyrimidine kinase, theenzyme required for the
phosphorylation of ACG (11, 25), this enzyme
activity was measured directly in the extracts whichwere employed in the DNA polymerase assay. Viral deoxypyrimidine kinase activity (units per microgram of protein) was 4.4, 4.0,
and 3.2 for the HSV-2WT-, R6-26-,and R6-34-infected cell extracts, respectively, in the
ab-senceofACG. In thepresenceofACG(20 ,uM),
7.2, 6.8, and 8.0 U/,ug of protein were detected
forthe HSV-2 WT-,R6-26-, and R6-34-infected
cellextracts,respectively. TheACG-treated
cel-lular extracts exhibited a twofold increase in
thymidine kinase activity in allcases. Since
al-kaline DNase activity could interfere with the template in the DNA polymerase assay and
perhapsaccountforapparentdifferences in viral DNA polymerase activity induced by different viruses, the alkaline DNase activity induced by therecombinant viruseswasalsodetermined. It
had previously beenshown, however, that under the assay conditions employed here an
oligo-(dG)12-81
poly(dC) templatewasnothydrolyzedby alkaline DNase (16). Alkaline DNase activity
wassimilar in allextracts,and theextracts
pre-pared with R6-26 and R6-34-infectedcells inthe
presenceand absence of ACG exhibited identical
specific activities. The activities of these two viralspecific enzymes appearedtobe identical
incellsinfected with theserecombinant viruses,
and it isunlikely thatthey playanysignificant
role in the differencesobserved in the viral DNA polymerase activities induced by these
recom-binants.
DISCUSSION
Inthisstudywehaveemployed
HSV-1/HSV-2 intertypic recombinants with well-character-izedgenomestructures todemonstrate thatthe mutations which confer resistance of HSV to ACG or PAA produce an altered HSV DNA
polymerase activity which exhibits resistanceto three inhibitors of DNApolymerase.
Two recombinants, R6-34 and R6-26, which containHSV-1 DNA insertions withinthe DNA polymerase locus ofanotherwise HSV-2genome
were selected forfurther analysis and
purifica-tion ofthe viral DNApolymerase. R6-34hasan
HSV-1 DNAinsertion extending frommapunits
39.6to42.8 including the region ofuncertainty and isACGr and PAAr. The R6-26recombinant containsanHSV-1 DNAinsertion startingfrom
the same HSV-1 BglII i-d site on the left
ex-tending from map units39.6 to 41.0, including the region of uncertainty, and is PAAS, and ACGS. The mutations in HSV-1 DNA which
confer resistancetoPAAandACGinthe R6-34
recombinant must be contained in the DNA
sequencesdefinedbymapunits41.0 to 42.8.By
analysis oftwoother
recombinants,
R6-19 andR6-30,these limitscanbefurther narrowed to
the DNA sequences defined
by
the HSV-2BamHIh'-j' sitetotheHSV-1Kpnx-csite
(map
units41.0 to41.8) or 1.3kilobase
pairs (5,
6,8).
Thisissufficientgenetic informationtoencode
for an amino acid sequence of 42,000 daltons
either as a separate
polypeptide
or part of alargerprotein. This
region
of DNAsequencesiswithin the
mapping
limits of the HSV-1(Kos)
tsD9 mutation
defining
the structural geneforHSV DNA polymerase (5) and is
likely
toin-clude the DNA sequence
defining
the activecenterof the HSVDNA
polymerase.
These two recombinants induce identical
amounts of
thymidine
kinase and alkalineDNase activityin thepresence and absence of
ACG (8), and the differences in
sensitivity
toACG andPAA cannotbe
explained
byadiffer-ence in the activities of these two enzymes.
These two recombinants induce identical
amountsof
ACG-TP,
theactivecytoplasmic
in-hibitor ofviralDNApolymerase,inthepresence
ofACG. TheR6-34recombinantinducesaviral
polymerase which is resistant to
ACG-TP,
whereastheR6-26recombinant and theHSV-2
WT induce DNA
polymerase
activities whicharesensitivetoACG-TP.
The
purified
DNApolymerase
inducedby
thesetworecombinants and the
parental strains,
HSV-2 ts6 (strain
HG52)
and HSV-1 strain 17(ts+,paar_j),possessasimilar molecularweight
of 142,000-144,000onsedimentationin
glycerol
density gradients. The
polymerase
activitieswere stimulated by high salt, inhibited by
N-ethylmaleimide,andpossessed the properties of
HSV DNA polymerase. The peak fractions of
the
purified
viral DNA polymerase wereem-ployed to testfor the sensitivity of HSV DNA
polymerasetoPAA, and theviral DNA
polym-erasesinducedbyR6-34 andthePAAr_1parent
wereresistanttoinhibition by PAA,possessing
ahighKi.
Partially
purifiedviralpolymeraseswerealsotested for sensitivity to inhibition by the four didNTP's. Thesedideoxy analogshave been in-structiveregardingthe mechanism of polymeri-zationbytheEscherichiacoli DNApolymerase, Pol I (2). The didNTP serves as polymerase substrate toelongateaDNAchainby one
resi-due. Achain terminated with such a
dideoxy-nucleoside is inert to further elongation and
relatively inert to exonuclease action at the
primer terminus or to attack by PPi. The
didNTP,
lacking
a 3'-OH group vital for chaingrowth, is still boundtothe dNTP bindingsite
on November 10, 2019 by guest
http://jvi.asm.org/
on the enzyme with the same
affinity
as thenatural triphosphate
and formsapolymerization
complex
protecting
theprimer
terminus fromnucleolytic attack. With theHSVDNA
polym-erases induced by the R6-26 recombinant and
theHSV-2ts6 parent,the didNTP'sact as
effec-tive
competitive inhibitors,
revealing
lowKi
val-ues(Table 3).Itisof interest that theapparent
Ki
of the human leucocyte polymerase fordidTTPis 9.6,uM (1), avalue ingeneral
agree-ment for the
Ki
values of the sensitive HSVDNA polymerases for didNTP's (14.7 to 22.6
,M).
The viral DNA polymerase of the R6-34
re-combinant orthe HSV-1
PAAr_i
parent,how-ever, wasmarkedly resistanttoinhibition by all
four didNTP's. Thissuggeststhat themutation
which confersresistance of these viral
polymer-ases to ACG-TP may confer resistance to all
four didNTP's as well. The data presented in
Fig. 2 indicating the effect of ACG-TP in the
cellularextract oninhibiting dGTP
polymeriza-tionby the viral polymerases of26 and
R6-34with the oligo(dG)12-18-poly(dC) template is
instructive concerning the role of ACG-TP in
binding to the viral polymerase. ACG-TP has
previously been shownto bind to the HSV DNA
polymerase and act as a competitive inhibitor
(11, 26). The R6-34 DNA polymerase is not
affected by the presence of ACG-TP and is
probably not bound by the inhibitor, whereas
the DNA polymerase ofR6-26 ismarkedly
in-hibitedby thepresenceofACG-TP, presumably
bybindingtothe viralpolymerase (Fig.2B and
C). Since the didNTP bindstothetriphosphate
binding siteontheE.coli PolI enzyme,and the
HSV DNA polymerase presumably also
pos-sessessuchasite,amutationintheDNA
polym-eraselocusonHSV which resultsinpreventing
anyanalog lacking a3'-OH groupfrom binding
with the same affinity as the natural
triphos-phatetothetriphosphate binding sitemayallow
polymerizationtoproceed. This mutation could
result in a more stringent
recognition
by thetriphosphate
binding
site of the enzyme andcould result in a failure to
incorporate
com-pounds
lacking
a 3'-OH group. Since thepro-posed site of action ofPAA onthe HSV DNA
polymerase is at the pyrophosphate exchange
site,amutation whichconfersresistance to PAA
and ACG-TP and didNTPsuggests that the
PPi
exchange site is very
closely
adjacent to thetriphosphate bindingsite in the enzyme. These
results provide additional supportfor the
pro-posalthat HSV DNA polymerase conforms to
the model of DNA polymerase advanced by
Kornberg,
i.e.,that DNApolymerase possessesanactive centercontainingmultiple sites, each
with adifferent function (18). Amutation
pro-ducinganalteration inone function of the active
center mayaffectthe others aswell.It has been
shownthat thesefunctionsare closelylinkedin
theE. coli PolI enzyme, and aconsequence of
chain terminationby didNTP's is the inhibition
of
PPi
exchangeand exonuclease action (2). InHSV DNA polymerase, the 3'--5' exonuclease
and
PPi
exchangefunctions may becloselyas-sociated because PAA, aninhibitorof
pyrophos-phateexchange,inhibits
3'-.5'
exonucleasefunc-tion to a similar degree (17). Unlike didNTP's
orACG-TP,however, PAA does not get
incor-porated into DNA, and it does not serve as a
substrate forHSVDNApolymerase (3, 22).
An alternative explanation for the observed
resistance of the R6-34 and PAAr_1 DNA polym-erases to inhibition by didNTP is that these
altered polymerasespossess anenhanced ability
toexcisethese analogsby the
3'-.5'
exonuclease,or"editing,"function of HSVDNA polymerase.
After incorporation of an altered analog, the
3'- 5' exonuclease function catalyzes the
re-moval of3'-terminalnucleosides from the primer
templateas
well
asthe template-dependentcon-version of dNTP's to monophosphates (17). A
DNApolymerase doesnot require such an
exo-nucleasefunction to be resistanttothe
inhibi-toryeffects of didNTP'sasbotheucaryotic
po-lymerases of CV-1 cells(10)and leukocytesfrom
patients with human myelogenous leukemia (1)
areresistanttoinhibition by theanalogdidTTP.
The cellular a polymerase from myelogenous
leukemia cells is sensitive to inhibition by
didTTP, however (1). Since neither the
eucar-yotica nor
,8
polymerasepossessesexonucleasefunction,
and sincethey differ in their sensitivitytodidTTP, the absenceof the exonuclease
func-tion doesnotexplain resistance. The mechanism
of resistancetodidTTP exhibited bya
polym-erase most
likely
is duetoitsability
todiscrim-inate between didTTP and dTTP,
preventing
incorporation
of the didTTP.A
potential
objection
to the use ofhybrid
genomesfor the purpose of
genetic analysis
ofherpes viral functions is that the recombinant
genomewill
produce
newrecombinantpolypep-tideswhichare soaltered from
parental proteins
that conclusions about
parental
functions arenotvalid. This appearstobean
unlikely
possi-bilityinthepresent case,
however,
becausethealtered resistanceof the DNA
polymerase
of theR6-34recombinant isobserved in the
parental
HSV-1 PAA-r virus as well. The
apparent
Ki
values for thedidNTP'sareverysimilar for the
DNA
polymerase
of the R6-34recombinant andthe HSV-1 PAAr_1 parent
(Ki,
135.1 and 122.51tM,
respectively).
Inaddition,
theKi
values foron November 10, 2019 by guest
http://jvi.asm.org/
756 KNOPF, KAUFMAN, AND CRUMPACKER
the DNA polymerase of the R6-26 recombinant and the HSV-2 ts6parentarealsoverysimilar,
andthesetwostrainsare sensitivetoPAA and ACG. The molecular weights of all four viral DNApolymerases are identical in the glycerol
gradient analysisandarethesameasthe molec-ular weight of the HSV-1DNApolymerase (17, 24).
The HSV-1 sequences which definethe
mu-tations for acgr and paar map in equivalent positions in both HSV-1 and HSV-2 (5, 6, 8). The type-specific sensitivity toACG in HSV-1 and HSV-2 canbe transferred separatelyfrom
theacgr mutation (8).Theseargumentssuggest that the viral functions in the parentalstrains
are represented faithfully in the recombinants
and that the recombinantgenomesprovide
use-fulprobes for theseparationandanalysisof viral
genefunctions.
Thisreportillustrates theutilityofcombining finestructuremappingofthe HSV DNA
polym-eraselocus withtheuseofspecificinhibitorsof
DNApolymerase functionstoseparatethe func-tions of the HSV DNA polymerase. This is a
goal which has previously been realized only withprocaryotic polymerases.Indefininga
1.3-kilobase-pair region of DNAsufficient to code for an amino acid sequence of42,000 daltons, which contains mutationstothree DNA
polym-eraseinhibitors ofdissimilarstructure,wehave
defined theregion coding for the active center of theHSV DNApolymerase molecule. PAA,a
PPi
analoginhibitingPPiexchange(19); didNTP analogs lacking a 3'-OH group and shown to bindtothetriphosphate binding siteonE. coliPol I (2); and ACG-TP, a compound which serves as asubstrate for the HSV DNA
polym-erase,isacompetitive inhibitor ofdGTP (11, 12,
26), and hasmanyfeatures incommonwith the
didNTP's,actonseparatefunctions inthe HSV
DNApolymerase; all inhibit the polymerization reaction. Whetheronemutationorthree
sepa-rateonesaccountforthe HSV DNA polymerase
resistancetothesecompounds, thefact that the resistancemutationsmapin suchasmall region
ofthe DNA provides supportthat this region definesanactive centerfor the HSV DNA
po-lymerase. Kornberg has describedanactive
cen-ter as a specially adapted polypeptide surface
whichrecognizes and accommodates several
nu-cleosidestructures(18), and thesestudies define such a region on the HSV DNA polymerase
polypeptide.IftheHSV DNA polymerasehasa
molecularweight of140,000to150,000,the
larg-estlimits of this active centerwould be molec-ularweight 42,000, aboutathird of theenzyme
molecule. Further definition and subdivisionof this region will require similar studies on
mu-tants and recombinants which separate resist-ancemutations to inhibitors of HSV DNA po-lymerase.
ACKNOWLEDGMENTS
Wegratefullyacknowledge the excellent technical assist-anceofMaryMurphy inpreparingthe virus stocks.
C.S.C. is therecipient of Public Health Service Research CareerDevelopment award 5-K04-CA--139 from the National CancerInstitute. K.W.K. is therecipient of aEuropean Sci-enceExchangefellowship from the Royal Academy of Science, UnitedKingdom,sponsoredbytheDFG,FederalRupublicof Germany. E.R.K. wassupported by Public Health Service grants CA16751 and CA26195 from the National Cancer In-stitute.
LITERATURE CITED
1. Allaudeen, H. S. 1980. Inhibition ofdeoxyribonucleic acidpolymerases of human leukemicleucocytesby 2',3' dideoxythymidine triphosphate.Biochem. Pharmacol. 29:1149-1153.
2. Atkinson, M. R., M. P.Deutscher,A.Kornberg,A. F. Russell, and J. G. Moffatt.1969.Enzymaticsynthesis of deoxyribonucleic acid. 34. Termination of chain growth bya2',3'-dideoxyribonucleotide. Biochemistry 8:4897-4904.
3. Bolden, A., J.Aucker,and A.Weissbach.1975. Syn-thesisofherpessimplex virus, vaccinia virus and ade-novirus DNA in isolated HeLacell nuclei. 1. Effect of virus-specific antisera andphosphonoacetic acid. J. Vi-rol. 16:1584-1592.
4.Bollum, F. J.1959.Thermal conversion ofnon-priming deoxyribonucleic acidtoprimer. J. Biol. Chem. 234: 2733-2734.
5. Chartrand, P.,C. S.Crumpacker,P.Schaffer,and N. M.Wilkie. 1980.Physical and genetic analysis of the herpes simplexvirus DNApolymeraselocus.Virology 103:311-326.
6. Chartrand, P.,N. D.Stow,M. D.Timbury,and N. M. Wilkie.1979.Physicalmappingofpaa' mutations of herpes simplex virus type1 and type2by intertypic markerrescue.J. Virol.31:265-276.
7.Coen,D.,and P. Schaffer.1980.Two distinctloci confer resistancetoacycloguanosinein herpes simplex virus type1.Proc.Natl. Acad.Sci. U.S.A. 77:2265-2269. 8. Crumpacker, C. S., P. Chartrand, J. H.
Subak-Sharpe,and N. M.Wilkie. 1980. Resistance of herpes simplexvirustoacycloguanosine-geneticandphysical analysis. Virology105:171-184.
9. Crumpacker,C.S.,L. E.Schnipper,J. A.Zaia, and M. J. Levin.1979.Growthinhibitionby acycloguano-sine ofherpes-viruses isolated from human infections. Antimicrob. AgentsChemother. 15:642-645. 10.Edenberg,H.J., S. Anderson, and M. L. De
Pam-philis. 1978. Involvement of DNA polymerase a in simianvirus 40 DNA replication. J. Biol. Chem. 253: 3273-3280.
11.Elion,G. B., P. A. Furman, J. A. Fyfe, P. De Miranda, L.Beauchamp,andH. J.Schaeffer.1977.Selectivity ofaction of an antiherpetic agent, 9-(2-hydroxyeth-oxymethyl)-guanine.Proc.Natl. Acad. Sci.U.S.A. 74: 5716-5720.
12. Furman, P. A., M. H. St. Clair, J. A. Fyfe, J. L. Rideout, P. M. Keller, and G. B. Elion. 1979. Inhi-bition ofherpessimplexvirus-induced DNA polymerase activity and viralDNA replication by 9-(2-hydroxy-ethoxymethyl) guanine and its triphosphate. J. Virol. 32:72-77.
13. Hay,J., and J. H.Subak-Sharpe. 1976. Mutants of herpessimplex virus types 1 and 2 that are resistant to phosphonoacetic acid induce altered DNA polymerase activities ininfectedcells.J.Gen. Virol.31:145-148.
on November 10, 2019 by guest
http://jvi.asm.org/
14. Honess, R. W., and D. H. Watson.1977.Herpes simplex virus resistance and sensitivitytophosphonoacetic acid. J.Virol. 21:584-600.
15.Jamieson, A. T., G. A. Gentry, and J. H. Subak-Sharpe. 1974.Inductionofboththymidine and deox-ycytidine kinase activity by herpes viruses. J. Gen. Virol. 24:465-480.
16.Jamieson, A. T., and J. H. Subak-Sharpe. 1976. Herpes simplex virus specifieddeoxypyrimidine kinase and the uptake ofexogeneousnucleosides by infected
cells.J. Gen. Virol. 31:303-314.
17.Knopf, K. W.1979. Properties of herpes simplex virus DNApolymerase andcharacterization of its associated exonucleaseactivity. Eur. J. Biochem. 98:231-244. 18.Kornberg, A. 1969. Activecenterof DNApolymerase.
Science163:1410-1418.
19. Leinbach, S. S.,J. M.Reno, L.F.Lee,A. F. Isbell, andJ. Boezi. 1979. Mechanism ofphosphonoacetate inhibition ofherpesvirus-induced DNA polymerase. Biochemistry 15:426-430.
20. Lowry,0.H.,N.J.Rosenbrough, A. L Farr, and R.
C.Randall. 1959. Protein measurement withaFolin phenolreagent.J.Biochem. 193:265-275.
21. Macpherson, I.,and M.Stoker. 1962.Polyoma trans-formation of hamster cell clones-aninvestigation of geneticfactorseffectingcellcompetence.Virology 16: 147-151.
22. Mao,J.C.-H., and E. Robinson.1975.Mode of
inhibi-tion of herpessimplex virus DNA polymeraseby phos-phonoacetate.Biochemiistry 14:5475-5479.
23. Morrison, J. M., and H. M. Deir.1968. Anew
DNA-exonuclease in cellsinfected with herpes virus: partial purificationandpropertiesoftheenzyme.J.Gen. Virol. 3:337-347.
24. Powell, K.L.,and D. J. M. Purifoy. 1977.Nonstructural
proteins of herpes simplexvirus. I.Purification ofthe induced DNA polymerase. J. Virol. 24:618-626. 25. Schnipper, L. E., and C. S. Crumpacker. 1980.
Resist-anceofherpessimplex virustoacycloguanosine: the role of viral thymidine kinase and DNA polymerase loci. Proc. Natl. Acad. Sci. U.S.A. 77:2270-2273. 26. St.Clair, M. D., P. A.Furman,C. M.Lubbers, and G.
B.Elion. 1980. Inhibition of cellular and virally induced deoxyribonucleic acid polymerases bythetriphosphate ofacyclovir. Antimicrob. Agents Chemother. 18:741-745.
27. Timbury, M. D., and L. Calder. 1976. Temperature-sensitive mutants ofherpes simplex virus type 2: a
provisionallinkagemapbasedonrecombination anal-ysis. J.Gen. Virol. 30:179-186.
28. Weissbach, A. 1975. Vertebrate DNA polymerases. Cell 5:101-108.
29. Weissbach, A., S.LHong, J. Aucker,and R.Muller.
1973.Characterization of herpes simplex virus-induced deoxyribonucleicacidpolymerase.J.Biol.Chem.248: 6270-6277.