0022-538X/92/010489-07$02.00/0
CopyrightC 1992, American Society for Microbiology
EBNA1 Can Link the Enhancer
Element
to
the
Initiator
Element of
the
Epstein-Barr Virus Plasmid
Origin of
DNA
Replication
TIM MIDDLETON* AND BILL SUGDEN
McArdleLaboratory forCancer Research, UniversityofWisconsin,
1400 UniversityAvenue, Madison, Wisconsin53706 Received29July1991/Accepted 8 October1991
The plasmid origin of DNA replication ofEpstein-Barr virus, oriP, is replicated once per cell division, employing cellularreplication machinery and onlyoneviral protein.Tounderstandhow replicationfrom this
origin is initiated andregulated,wepurified this viral protein, EBNA1. EBNA1wasexpressed inCV-lpcells
by using aninfectious simian virus 40 vectorcontaining the EBNA1 gene. Itwas purified in two chromato-graphic steps to apparent homogeneity. The purified protein is capable of supporting transcription of the luciferasegenefromareporterplasmid carrying the FR enhancerelementtowhich EBNA1 binds.EBNAldoes nothave oriP-dependentATPase activity, indicating that it doesnotcarryoutanenergy-dependentstepin the initiation of DNA replication.However,EBNAldoes mediateanassociationbetween thetwoelementsoforiP. We measured thisassociation by bindingone of the elements, the enhancer element, to a solid matrix and
measuring retention by thiselement of the otherone,the initiator element, in the presenceofEBNAl. This retention is specificforDNA fragments containing EBNAl-binding sites.EBNA1 thuscanlink thetwoelements of theorigin, providingalocally highconcentration of EBNA1atthesiteof initiation of DNA replication. We proposethat thisassociationis important either (i)toaffect DNAstructure toallowacellular helicasetoinitiate DNA strand separationor(ii)tobindreplication proteinstobring themtothe origin of replication.
DNAreplication requires careful coordination of the
en-zymatic steps of replication with other aspects of cell
divi-sion. Studies usingprokaryotic replicons have identified the
primary enzymatic activities required for DNA replication
and, in some cases, mechanisms by which replication is
regulated (21). More recently, genetic studies using herpes
simplex virus and reconstructions of replication activity in
vitro using simian virus 40 (SV40) and adenoviruses have
demonstrated that the enzymology of DNA replication is
well conserved from bacteriato mammals (6, 31). However, the regulation ofDNA replication within the cell cycle in
mammalian cells isnot well understood.
In mammalian cells, replicons must be copied only once per cell cycle. There have been two impediments to deter-mining how this regulation occurs. (i)Those viralorigins of replicationused toidentifythe
required
enzymatic functionsare derived from
lytic
viruses and are, by their nature, notlimitedto oneroundofreplicationpercellcycle.
(ii)
Eukary-otic chromosomal origins ofreplication
have notbeenmade tofunction invitro; incontrasttoorigins
fromSaccharomy-cescerevisiae, mammalian chromosomal
origins
have been studiedasplasmids in vivo withonly
limited success. Until the features of these chromosomalorigins
that limit their study in isolationareidentified,
thesereplicons
willprovide
cumbersome models both forthe
study
ofthe initiation ofDNAreplication and for itscellcyclecontrol.
Latently replicating viruses
bridge
the gap betweenlytic
viruses and chromosomal
origins
ofDNAreplication.
Thebest studied of these virusesare
papillomaviruses
andlym-photrophic herpesviruses.
Epstein-Barr
virus(EBV)
is alymphotropic
herpesvirusthatimmortalizesBcellsoninfec-tion.
Replication
of the viral DNA issubject
to the sameconstraintsaschromosomalDNA
during
onephase
of its life cycle. EBVisdistinguished
from chromosomalreplicons
in*Corresponding author.
that its origin has been identified andfunctions in plasmids
(34). EBV has a linear genome ofapproximately 172 kbp which iscircularizedafter infection and is maintained in the nucleus as aplasmid at a copy number of between 5 and 500
(30). While there is considerable variation incopy number betweenclonesof infectedBcells, oncethe copy numberis reached at some early stage afterinfection, itis stable for many generations thereafter. Furthermore, density shift experimentshave demonstrated that EBV DNAisreplicated
once per cell cycle (1, 36).
Genetic analyses of the plasmid origin of EBV DNA
replication (oriP)havedemonstratedthat less than 800bp of
EBVDNAisneeded toformafunctionalreplication origin. ThisDNA is divided intotwoelements, which inthe virus are separated by about 1,000bp of
intervening
DNA (27).Both elements, the family of repeats (FR) and the dyad
symmetry region (DS), consist of multiple
copies
of arecognition site for viralprotein EBNA1 (25), which is the
only viral protein
required
for DNAsynthesis
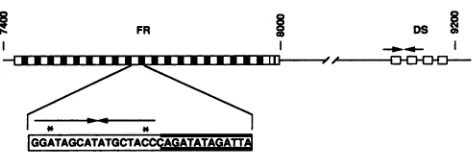
from oriP(Fig. 1; 16,37). DNA
replication
initiatesat or nearthe DS element(10), while FRisanaccessory elementrequired
for DNAreplication (27).Two features of
particular
interestinunderstanding
oriPfunction are the interactions ofEBNA1 with other
replica-tion proteins and the mechanism
by
which the FR and DS elementsinteracttoformafunctionalorigin.
Tostudy
thesefeatures,
wepurified
EBNA1 from cells in which EBNA1and oriPfunction. We demonstrated that
purified
EBNA1 isbiologically
activeby
measuring
itsactivity
in vivo afterintroduction of the
protein
back into mammalian cells.Purified, functional EBNA1 does not have detectable ATPase
activity,
indicating
that it isnotlikely
toperform
theenergy-dependent
strandseparation
thatrepresents anearly
enzymatic
activity
in DNAreplication.
Itdoes,
however,promoteanassociationbetween FR andDS that is
likely
tobe
required
for initiation of DNAreplication.
489
on November 10, 2019 by guest
http://jvi.asm.org/
FR
-1p
I*-*A
FIG. 1. Structure of oriP. FR and DS are indicated as boxes correspondingto therepeat units. Thepositionof oriPontheEBV
genomeisgiven bythenucleotide numbersof thesequenceofBaer
et al. (4). The arrows indicate inverted repeats. The lower part showsa consensusrepeatsequence.The asterisksmarkmismatches
intheinvertedrepeattowhichEBNA1binds(3, 13).
MATERIALS AND METHODS
Plasmids. The basepairnumbersgivenfor EBVarethose describedbyBaer et al.(4).p1010 andp304contain the DS
(EBV bp 8992 to 9132) and FR (EBV bp 7315 to 8191)
elementsinserted intotheBamHIsites ofpBKSandpSP65, respectively. p972 contains oriP (EBV bp 7333 to9257) in the
HincII
and SphI sites ofpUC19. p896isa derivativeof pBluescriptKS'
with a duplex oligonucleotide with the sequence AGCTTATCTATATCTGGGTAGCATATGCTA TCCTAATCTAGGATCCAinserted at theHindIll site. The binding siteforEBNA1 isunderlined.p1013 (tk-luciferase) was constructed by removing from
pA2(-260/-87)tkCAT8+
(14) the HindIII-to-BamHIfrag-ment containing DNAfrom thepromoter region ofa vitel-logeningeneandreplacingthechloramphenicol
acetyltrans-ferase gene on a BgIII-to-HpaI fragment with the firefly
luciferase gene on a HindIII-to-HpaI fragment from pSVL (8). p985 (FR-tk-luciferase) contains the FR element on a BamHIfragment fromp304 inserted into theBamHI siteof
p1013.
The SV40-EBNA1 expression vector has been described previously (11). Briefly, aBamHI K fragment containinga
deletionof mostof theglycine-glycine-alanine repeat region
of EBNA1 was inserted into the XhoI site of plasmid pSVEpR4. The deleted region is not essentialfor EBNA1-mediatedactivationoftranscriptionorreplication (35).EBV
bp 109,982 to 112,065 were replaced by aBamHI linker to give p398Y. Digestion of this plasmid with BamHI gave a fragmentofpackageablesizecontainingtheSV40 originand
early region,withtheEBNA1 genereplacingthelateregion.
The EBVsequencesincludedarefrombp107,567to109,982
and 112,065to 112,622.
The plasmid used to complement the deletion in p398Y
contained SV40 DNA with a deletion ofbp 4974 to 5046,
whichcreatesanout-of-framedeletion intheT-antigengene
(provided by JanetMertz).
Infectionand harvestingofCV-lpcells. Stocksofviruses
were obtained by introducing the complementing viral
DNAsintoCV-lpcells byDEAEtransfection(17). At 96 h later, the cells were harvested and virus was recoveredas
described in reference 18. These virus stocks were used to infect CV-lp cells for purification of EBNA1. The ratio of
virustocells touseforinfectionwasdetermined by titrating
thevirus onCV-lp cells, measuring the amountof EBNA1 produced by an enzyme-linked immunosorbent assay (see
below),andusingthattiterwhich maximizedtheamountof
EBNA1detected.
Infections for isolation of EBNA1 were done on
15-cm-diametertissueculturedishes.CV-lpcellsweregrownto50
to 80%confluence inDulbecco'smediumplus 10% neonatal
calfserum. Medium was
removed,
and viruswas incubatedwith the cellsin 3 mlof mediumfor1 h.
Thirty
milliliters of medium wasadded,
and dishes were incubated until about10% ofthe cells started to detach. Medium was
aspirated
fromthe
dishes,
and cellswerescraped
offtheplates
intwovolumes of buffer D
(20
mM HEPES[N-2-hydroxyethylpi-perazine-N'-2-ethanesulfonic
acid]
[pH 8],
1 mMEDTA,
1%Triton X-100, 0.02% sodium
azide,
30 mM sodiumPP1,
50 mM NaF, 1 mM sodiumorthovanadate,
and a mixture ofprotease inhibitors
[20]).
The cells were stored at -70°C until used.Purification and monitoring of EBNA1.
CV-lp
cells ex-pressing EBNA1 werelysed by being
passed
twicethrough
a French press at 800 lb/in2 in two
packed
cell volumes ofbuffer D. Ammonium sulfatewas added to
30%
saturation,
and lysateswere left onice for 1 h and spunat
100,000
x g for 1 hat4°C. Supernatantswerediluted in bufferDtogive
an ammonium sulfate concentration of0.4 M.Supernatant
from 109 cellswas
applied
to a10-mlheparin-agarose
column and washed with 10 column volumes of0.4 M ammoniumsulfate, andtheEBNA1-containing fractionwaselutedwith 3 column volumes of0.7 M ammonium sulfate. The eluate was diluted to 0.3 M ammonium sulfate and applied to an
affinity column madeoftheBamHI FR
fragment
ofp304
as described by Kadanoga andTjian
(12). The column waswashedwith 10column volumesof buffer E (20 mM HEPES
[pH 8.0], 1 mM EDTA, 1 mM dithiothreitol)
plus
0.6 MNaCl, and EBNA1 was eluted with 2 column volumes of buffer E plus 2 M NaCl. The eluate was concentrated
by
using carboxymethyl cellulose (Aquacide IV;
Calbiochem)
asrecommendedbythemanufactureranddialyzed in buffer
E with 0.2 MNaCl and 15% glycerol. The
purified
protein
was storedin this buffer at
-70°C.
Theelution characteristics of EBNA1 weremonitored
by
anenzyme-linked immunosorbentassay.Threefold dilutions
of 1 ,l of the protein-containing solution were bound to
nitrocellulose in 100 ,l of 10 mM sodium phosphate (pH
7.2)-S150
mMNaCI-20 ,ug of bovine serum albumin per ml. Twofold dilutions of 20 ng of a lambdacro-EBNA1-,-galactosidase fusion protein (30) were similarly bound. The amounts of EBNA1 in purification fractions were
deter-mined by densitometric scanning of immunoblots probed
with antibodies made to the fusion protein (30).
The purity of the protein was determined by sodium
dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis,
followed by staining ofthe gel with Coomassie blue. The concentration of purified EBNA1 was determined by flu-oraldehyde assay as recommended by the manufacturer
(Pierce Chemical Co.).
DNA-binding assay. The DNA used was either p896 or the oligonucleotide used to construct p896. Twenty-five femto-moles of end-labelled (28) DNA was bound to protein in 50 ,l of 20 mM HEPES (pH
8.0)-i
mM EDTA-1 mM dithio-threitol-0.2 M NaCl-50 ,ug of bovine serum albumin per ml for 10 min at 20°C. The samples were filtered throughnitrocellulose by using a dot blotting apparatus from Schlei-cher & Schuell and washed with 400
Rl
of 20 mM HEPES (pH8.0)-i
mM EDTA-1 mM dithiothreitol-0.2 M NaCl. Dots were cut out and counted with a scintillation counter. Introduction of purified EBNA into mammalian cells. EBNA1 was incubated with luciferase reporter plasmids underthe conditions used for the DNA-binding assay. TheDNA-EBNA1 mixture was introduced into 143 cells by electroporation by using a rise time of 8 ,us, a voltage peak of 760 V, and a time of 30 ms for decay of the voltage to one-third of its peak (15). Extraction of luciferase from the
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.67.303.71.147.2]cells andmeasurementofluciferaseactivitywereperformed aspreviouslydescribed (5).
ATPaseassay. ATPase activity was measured by absorp-tion of unhydrolyzed ATP by charcoal. Reacabsorp-tionscontained
200 mM KCl, 10 mM MgCl2, 50 ,ug of bovineserumalbumin
perml, 1 mM EDTA, 0.2% Triton X-100, 3% glycerol, 1 mM
dithiothreitol, 0.1 to 1 1xCi of [-y-32P]ATP, 50 mM HEPES (pH 8.0) (unless otherwiseindicated),1 pmolof EBNA1, and 0.2 pmol of DNA (unless otherwise indicated). A 0.2-pmol sample of the DNA containing EBNAl-binding sites
con-tained 10 pmol of binding sites. After incubationat37°C for 20 min, 200 ,ul of a 20% (wt/vol) suspension of activated charcoal (Norit; Sigma Chemical Co.) in 10 mM sodium
PP-2%trichloroacetic acidwasadded andmixed for10min.
Thecharcoalwaspelleted, and 50,ul of thesupernatantwas
removedforscintillation counting.
FR-DS associations. FR bound toagarose resin was pre-pared as previously described (12). EBNA1 was incubated with 10 fmol of end-labelled DNA containing
EBNA1-binding sites by using the reaction conditions described for thenitrocellulose filter bindingassay.This mixturewasthen incubated with 10
RI
of resin (500 fmol of DNA) for 60minat 4°C. The resin was pelleted and washed twice for 20 mineachtime with 200 ,ul ofbinding buffer. The DS-containing DNAwaseluted by incubation for 10minat65°C in 20 ,ul of 10 mM Tris-HCl (pH 8.0)-i mM EDTA-0.1% SDS. The eluted material was electrophoresed on an agarose gel and visualized by autoradiography.
RESULTS
Construction ofanEBNA1 expression vector and purifica-tionof the protein. Expression of EBNA1 in EBV-positive lymphoblastoid cells is limited to about 5 x 104molecules percell(30). To facilitatepurification, apackageable
expres-sion vector containing the EBNA1 gene replacing the late
region of SV40 (provided by John Yates) was used. This
DNAwastransfected intoCV-lp cells along witha
comple-menting viral DNA containing a frameshift deletion in the
T-antigengene(provided by Janet Mertz)togeneratestocks ofinfectious virus. These viruses were then used to infect CV-lp cells for production of EBNA1. The infected cell populations typically contained approximately 106 EBNA1 moleculespercell, orabout 100 p,gof EBNA1 per 109 cells (datanot shown).
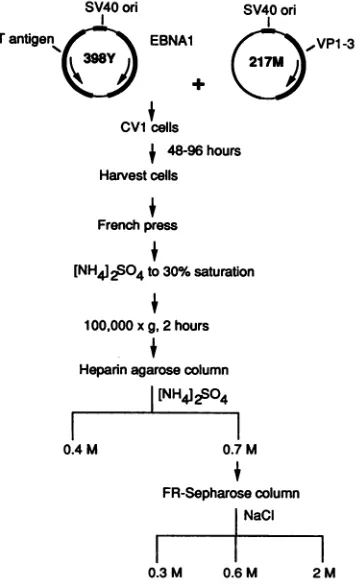
The scheme used to purify EBNA1 is shown in Fig. 2. Both protease and phosphatase inhibitors were required to maintainthe integrity of the protein through the first steps.
The purification required two chromatography steps, as
indicated inMaterials and Methods. Afterasinglepass over
theaffinitycolumn, EBNA1wastheonly band visibleonan SDS-polyacrylamide gel. A typical gel is shownin Fig. 3A. Immunoblotting of the purified material generallygave one
band that corresponded to the position of the Coomassie blue-stained band (Fig. 3B, lane 1). The exception to this
finding occurredinearly preparations inwhichphosphatase inhibitors were not used. In these cases, EBNA1 was
distributed among three to six bands spaced about 2 kDa apart, as shown in lane 2 ofFig. 3B. This observation is
consistentwith the interpretationthat the multiple forms of
EBNA1 detected resulted from its dephosphorylation after
thecells were harvested. The EBNA1 isolated in the
pres-enceofphosphatase inhibitors appearedtobehomogeneous, which indicates that its overexpression did not result in
heterogeneous phosphorylation of theprotein.
Binding of purifiedEBNA1 toDNA. EBNA1 binds
specif-SV40ori
Tantigen EBNAl
C
Cvi cells
SV40ori
1 ,VP1-3
f 48-96hours
Harvest cells
f
Frenchpress
f
[NH4]2504to30% saturation
100,000xg,2hours
Hepannagarosecolumn
[NH412S04
0.4 M 0.7M
FR-Sepharose column NaCI
0.3M 0.6M 2M
FIG. 2. Outline of the protocol used to express and purify EBNA1. Packageable SV40 vector398Y hasareplacement of its late region with the EBNA1 gene. 217M contains a frameshift mutation in theTantigen gene but supplies viral late gene products. Details of thepurificationaresupplied in Materials and Methods.
icallyto a 30-bpsequence thatis repeatedmultiple times in FRandDSoforiP. Retention of thisactivityin the purified
proteinwas tested by using a nitrocellulose binding assay.
Binding ofEBNA1 to 25fmol of eithera30-bp
oligonucleo-tidecontaining a consensusDNA-binding siteor a plasmid
containingonecopy of thisoligonucleotidewaslinear overa
10-fold range (Fig. 4). EBNA1 has been shown to bind to DNA as a dimer (2, 9), and it will be assumed here that a
dimer is the active form of EBNA1. These data show a
one-to-one correspondence between the moles of EBNA1
dimer added and the moles of probe DNA retained. This
binding was specificfor the EBNAl recognitionelement in that EBNA1 did not retain on nitrocellulose either the
parental plasmid lacking this 30-bp recognition site or an
oligonucleotide containing a recognition element for the estrogenreceptor(datanot shown).
Biological activity ofpurified EBNA1. FR acts as a
tran-scriptional enhancer when bound
by EBNA1,
both with heterologouspromoters andwithapromoterlocatedabout3kbp away in the EBV genome (26,
32).
This element sup-ports DNAreplication
with thesameflexibility
initsposition
and orientation as its
displays
insupporting transcription
(33). Furthermore, mutations in EBNA1 that affect DNA replication affect RNA
transcription (23, 35).
Weused the transcriptionalactivating
property ofEBNA1 and FRtotestwhether the
purified
EBNA1protein
wasbiologically
active.EBNA1
protein
bound to aplasmid
containing
FR,
the herpessimplexvirustype 1thymidine
kinasepromoter,andthe firefly luciferase gene was
electroporated
into cells.on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.349.529.76.368.2]A
Kd
97-68
-
43-
29-12
B
1
2
68-
43-FIG. 3. Gel electrophoresis ofpurified EBNA1. Samples were resolvedon an SDS-10%polyacrylamide gel. (A)Gel stained with Coomassie blue. Lanes: 1, supernatant from precipitation of an infected cellextractwith 30%(NH4)2SO4; 2, 1,ugof EBNA1 eluted fromthe DNAaffinitycolumn. Kd, kilodaltons. (B)Immunoblot of purified EBNA1. Lanes: 1, purification in the presence of phos-phataseinhibitors as indicated inMaterials andMethods;2, purifi-cation in the absence ofphosphatase inhibitors. Both lanes con-tainedapproximately 100ngofEBNA1.
These cellswere harvested 12 to72 hlaterandassayed for
luciferase activity.
We first tested whether EBNA1 remained bound to the DNAundertheconditionsusedforelectroporation. EBNA1
wasboundtotheDNAasdescribedearlier, diluted into the medium used for electroporation (but lacking serum), and
filtered through nitrocellulose. DNA retention was unaf-fectedby thistreatment, indicating that the DNA remained
boundby EBNA1 (datanotshown).
60
fmol EBNA1
FIG. 4. RetentionofDNA containingone EBNAl-bindingsite
on nitrocellulose after its incubation with purified EBNA1. A
25-fmol DNAprobesamplewasused foreachreaction.Theresults
shownarecombineddatafrom threeindependentexperiments using
either oftheprobesdescribed inMaterials andMethods.
The EBNA1-plasmid complex was then
electroporated
into 143 cells, an EBV-negative human osteosarcoma cell line. The effect ofthe ratio of EBNA1 to
plasmid
and thetime courseofaccumulation of luciferase
activity
areshown in Fig. 5. The amount of luciferase activity increasedlin-earily up to a fourfold molar excess ofEBNA1 dimer with respect tobindingsites,whereit reacheda
plateau (Fig.
5A).
Itis not known whetherthe molar excess ofEBNA1relative
tobinding siteswasneededtoachieve maximalsaturation of
binding sites or whether anincreased nuclearconcentration ofEBNA1 simply maintained
transcriptional
activation foralonger time. Luciferase activity peaked
approximately
2 days after transfection, although a significant amount of activity remained at 3 days (Fig.5B).
EBNA1 lacks ATPase activity. An early step in DNA replicationis energy-dependentunwinding ofthe DNAatthe origin. We determined whether EBNA1 was an
oriP-depen-dent ATPase. First, [32P]phosphate release from ATP was
measured in the presence oforiPDNAfrom severalstepsin the purification ofEBNA1 (Fig. 6A). The supernatantfrom
ammonium sulfate precipitation contained ATPase
activity;
however, the fraction from the heparin-agarosecolumn that
contained EBNA1 did not. Thus, as assayed here, there were no detectable ATPase activities that copurified with EBNA1 through the last two purification steps. The pres-ence of ATPase activity was notaffectedby the absence of
DNA in the assay (data not shown).
We also examined the starting material and the 0.7 M eluate from the heparin-agarose column for ATPase
activi-ties over a pH range of 5 to 9 (Fig. 6B). With the starting
material, there was substantial ATPase activity over the entire pH range, with a maximum at pH 8. Again, in the fractions eluted from the column containing EBNA1 there was no detectable ATPase.
EBNAI-EBNAl interactions promote an association be-tween the two elements oforiP. The genetic requirement of
two groups of DNA-binding sites for EBNA1-dependent EBV oriPreplication activity (27) led us toexamine whether these two elements form a complex with each other via a bridge of EBNA1. In preliminary experiments, purified
EBNA1 was added to p972 which had been digested such that FR and DS resided on separate DNA fragments. Asso-ciation between these two fragments was analyzed by elec-trophoresis in 1% agarose gels. Increasing amounts of EBNA1 shifted the labelled DNA fragments to the wells of thegel, although the mobility of fragments lacking
EBNA1-binding sites was unaffected (data not shown). Thus, we could not determine whether complexes between the DS-andFR-containing fragments had formed.
To circumvent this problem, we immobilized an FR-containing fragment on agarose beads and then determined whether the DS element, when bound by EBNA1, was retained on this matrix. We titrated the amount ofEBNA1 needed toretain the DSfragment on the resin. A 10-fmol DS fragment sample (40 fmol of binding sites) was incubated with 10 to1,000 fmol of the EBNA1 dimer, and this mixture was added to 10 ,ulof resin containing 500 fmol of the FR fragment (10 pmol of EBNA1-binding sites). The binding reaction was allowed to proceed for 1 h at 4°C and was washed twice with 20 volumes (each time) of binding buffer toremove the unbound DS fragment. Binding was detectable with addition of 30pmol ofEBNA1 and increased through-out the entire range of EBNA1 concentrations (Fig. 7A). When 1,000 fmol of EBNA1 was added, 40% of the DS fragment was bound to the resin. This binding was absent when the samefragment lacking the DS element was used as
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.80.287.75.286.2] [image:4.612.65.301.507.674.2]2 4 6 8 EBNA1/binding sites for FR-tk-Luc
B 5
4
-J
0
3
2
0
Ic
[image:5.612.72.545.77.215.2]Hours after transfection
FIG. 5. Transcriptional activationin vivo by purifiedEBNA1. PurifiedEBNA1 wasboundto reporterplasmids and electroporated into 143 cells, andextractsofthecellswereassayedfor luciferase activity.(A)Titration of EBNA1. A3-,ugsample ofreporterplasmidswasbound bya0.5-to8-fold molarexcessofEBNA1relativetobinding sites in FR-tk-luciferase. After24 h,cellswereharvested and assayed. (B) Time
course of luciferase expression. One microgram ofFR-tk-luciferase was incubated with afourfold molar excess ofEBNA1 relative to DNA-binding sites for EBNA1, electroporated into 143 cells, harvested, and assayed for luciferase activity atthe indicated times after electroporation. RLU, relative lightunit.
theprobe, when EBNA1 wasomitted from the reaction, or
with addition of polyclonal antibodiesto EBNA1 (Fig. 7B).
Thus, the FR element was able to retain specifically the
DS-containing DNA inareactionthatrequiredboth the DS element and EBNA1.EBNA1-mediated binding of DStoFR wasnotafunction of thestructureoftheDS element. When aplasmidcontaining justoneEBNA1DNA-binding sitewas
substitutedforthe DS fragment, itwas also retained by the FRelement(Fig. 7B).
DISCUSSION
While the replication origins of several mammalian lytic viruses have been studied intensively, our knowledge of origins thatarereplicatedoncepercellcycle is much more limited. The structural organization of oriP is similartothat ofanumberof prokaryoticorigins, in particular, origins that are understrict copy number control (21). It is not known whether this organization extends to mammalian chromo-somalorigins, althoughautonomously replicatingsequences derived fromyeastchromosomes do share this organization (22).
In common with these prokaryotic origins, oriP binds a
replicon-encoded protein that recognizes the repeat ele-ments that make up the origin. We purified oriP-binding
protein EBNA1 from mammalian cells. Thepurified protein retained biological activity, as Tmeasured by its capacity to stimulate a reporter gene when EBNA1 and a reporter
plasmid containing the FR enhancer and the firefly luciferase
gene wereintroduced intocells. Purified EBNA1stimulated
luciferaseactivity 200-foldoverthatseenin itsabsence. One uncertainty regarding this assay is the extent to which the stimulationof luciferaseactivity results fromanincreasein
transcriptional activation by EBNA1.AdditionofEBNA1to this reporter plasmid provides for attachment of up to 21
EBNA1 dimers possessing up to 42 nuclear localization signals (2, 27).These nuclearlocalization motifs maytarget
the electroporated DNA to a compartment of the cell in which it is transcribed efficiently. However, ifthistargeting
were the only mechanism by which EBNA1 stimulates
transcription of the reporter plasmid, then-introduction of
the reporter plasmid bound by EBNA1 into cells already expressingEBNA1 would result inanincrease in luciferase
activity relative to that caused by introduction of naked
DNA. We didnotdetect suchanincrease (datanotshown). This finding would be expected if the increase in luciferase activity observed (Fig. 5) resulted largely fromactivation of transcription and notsimply from targeting of the reporter DNAtothenucleus. Theamountof EBNA1 thatenterseach celluponelectroporation is likelytobe small relativetothe approximately 40,000 molecules already in these EBNA1-positive cells and thereforecannotcontributesignificantlyto thetranscriptional activation observed.
Purified EBNA1 lacksdetectable DNA-dependent ATPase activity (Fig. 5), which makes it unlikelytohaveanintrinsic helicase activity. EBNA1 purified from insect cells also lacks detectable helicase activity (9). These observations indicate that EBNA1 probably contributes functions tothe EBVplasmid replicon other than ATP-dependent unwinding of the DNA. Because localizedunwinding of the DNA bya helicase is likelyto be an early enzymatic activity required for DNAreplication,oneof theactions of EBNA1 maybeto bind a cellular helicase to the origin. The availability of purified EBNA1 allows this hypothesistobe tested.
Purified EBNAl can link the two cis-acting elements of oriP, FR and DS(Fig. 6). We imagine that this association
occursviainteractions ofEBNA1dimers boundtoFR with
those bound toDS, although wecannot ruleout the possi-bility thatasingle dimer is capableofbindingtositesonboth
molecules. The interaction measured herewas
intermolecu-lar. Intramolecular association betweenDS andFRhas also
beenvisualizedby electron microscoy (9a, 31a).
Anotable feature of the observed intermolecular
associa-tion was the relatively small number of EBNA1 molecules needed to mediate it. Forthe binding assay, EBNA1 was incubated with the DS element and then this mixture was incubatedwiththe immobilizedFR element. BindingofDS to FR was readily detectable when there was a 300-fold excessofFR-binding siteswithrespecttoEBNA1,and 40%
of theDSfragmentwasbound witha1:10 ratio ofEBNA1to binding sites. This indicates that a singledimer ofEBNA1
bound to FR is probably sufficient to mediate association
with DS boundby EBNA1 (Fig. 6A).A relatedphenomenon was first noted by Milman and Hwang (19), who used a fragmentof EBNA1 that includedthe carboxy-terminal 191 amino acids. This portion of EBNA1 was sufficient to associatetwoDNAfragments containing single high-affinity
consensus EBNA1-binding sites. Figure 6B confirms this
A 5
4
-J If)
0 3 2
0
on November 10, 2019 by guest
http://jvi.asm.org/
A
fmol
EBNA-1
dimer
00 0 0
O- -
M)-50 -1
40
-
30-
20-0.
4 5 6 7 8 9 10
pH
FIG. 6. ATPase activities detected during purification of EBNA1. ATPase activity was measured by incubation of protein with ATP andseparation of unhydrolyzed ATP from phosphate by absorption by charcoalasdescribed in Materialsand Methods.(A) ATPaseactivity associated with severalstagesofpurification. The fractionsassayedareindicated below thebargraph. Theamountof each fraction usedcorrespondedtotheextractfromapproximately 105, 105, and 4 x 105 cells for the 100,000 x gsupernatant (sup), heparin-agarose, and purified EBNA1 fractions, respectively, after correction forrecoveryof EBNA1ateachstep.(B) Effect of pHon
ATPase activity. Symbols: E3, material applied to heparin-agar-ose;*, material eluted from heparin-agarose.
finding and shows further that the lower-affinity sites which
composeDScanalsoformacomplex undertheconditions in
which the single higher-affinity sitewas bound.
This result indicates that not all of the multiple binding sites found in FRareneeded for EBNA1-mediated
associa-tion with DS. Wesuggest twoadditional propertiesthat FR
mayprovide this origin. One is that the multiple binding sites of FRbound by EBNA1 are needed tostabilizean
alterna-tive structure of the DS element bound by EBNA1 that is
obligatory for initiation of replication. Forexample, the DS element is theoretically capable of forming a cruciform structurethat extendsoverapproximately65bp. Chittenden etal. (7) have demonstrated that deletions that disrupt this potential cruciform but do not affect the EBNA1-binding sites render the DS element nonfunctional. Interactions of
FIG. 7. Associationof FR and DS elements via bound EBNA1. EBNA1wasincubatedwitha32P-labelled DNAfragment (3,152 bp) which contains the DS element, and this mixture was added to
immobilizedFR. Theretained DSfragmentwas eluted withSDS, electrophoresed,andexposedtofilm. Theresultingautoradiographs
areshown.(A) Titrationof the amounts ofEBNA1 needed to retain DS. The amounts ofEBNA1 added are indicated at the top. (B) RequirementforEBNA1- and DNA-binding sites for this associa-tion. Eachanalysiswasperformedinduplicatetoyieldtwo laneson
theautoradiogram. Probe DNAs: DS, plasmid containing the DS element;1 site, plasmid896 containingonecopyoftheconsensus
EBNA1-binding site; no sites, pBluescript KS' containing no binding sites. For EBNA1, treatments with added EBNA1
con-tained 100 fmol of the EBNA1 dimer. Foranti-EBNA1, DS and EBNA1 were incubated for 10 min, 1.5 ,ug of rabbit polyclonal anti-EBNA1 antibodieswasadded,andthe incubationwas contin-ued for an additional 20 min before addition of the mixture to
immobilized FR.
EBNA1molecules boundtoDSwiththoseboundtoFRmay
help stabilize suchastructure. Asimilar roleindistortingthe structureof lambda DNAnearitsorigin byits0proteinhas
been proposed (29).
Asecond candidatepropertyispromotion ofthe accumu-lation of cellular proteins needed to form a replication complexattheorigin by concentrating EBNA1 atthis site.
This effect could occur either by direct interaction of EBNA1withspecific replicationproteinsor,moregenerally, by bringingtheorigintoanuclear sitewhere theseproteins arelocalized.Thissuggestedpropertyof the FR element and EBNA1 would explain the apparently similar roles of EBNA1 inactivating transcription and replication. Multiple copies of EBNA1 localizedatFRwould activate transcrip-tion by forming heterologous complexes with other tran-scriptional activators, of viral or cellular origin. Similar predicted interactions form the basis for the looping model of
enhancer function (24). Given the similarity in the
charac-teristics of EBNA1 as anactivatoroftranscription andas a
0D C:
03
.0
C0
2
D
E~
0 CO C)
0 0
00)
B
.4.
c
41.
cx
In
-i
C. CM
De
:n1
f
I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.64.304.78.443.2] [image:6.612.318.555.81.349.2]componentof the replicationapparatus, anunderstanding of
howEBNA1 facilitates replication may
also
aid ourunder-standing ofhow transcriptional activators work. ACKNOWLEDGMENTS
We thank Julie Breister, David Anderson, and StephenHussey for technical assistance and Toni Gahn and Paul Lambert for comments on the manuscript. We thank Janet Mertz for providing plasmid 217Mand'JohnYates for providing plasmid 398Y.
This work was supported by Public Health Service grants CA-22443 and CA-07175 from the National Cancer Institute (B.S.), American Cancer Society grant IN-35-31-19, and a fellowship from the Cancer Research Institute (T.M.).
REFERENCES
1. Adams, A. 1987. Replication of latent Epstein-Barr virus ge-nomes in Raji cells. J. Virol. 61:1743-1746.
2. Ambinder, R. F., M. A. Mullen, Y. N. Chang, G. S. Hayward, and S. D. Hayward. 1991. Functional domains ofEpstein-Barr virus nuclear antigen EBNA-1. J. Virol.65:1466-1478. 3. Ambinder, R. F., W. A. Shah, D. R. Rawlins, G. S. Hayward,
and S. D. Hayward. 1990. Definition of the sequence require-ments for binding of the EBNA-1 protein to its palindromic target sites in Epstein-Barr virus DNA. J. Virol. 64:2369-2379. 4. Baer, R., A. T. Bankier, M. D. Biggin, P. L. Deininger, P.J. Farrell, T. J. Gibson, G. Hatfull, G. S. Hudson,S. C.Satchwell, C. Seguin, P.S. Tuffnell, and B. G. Barrell. 1984. DNA se-quence and expression of the B95-8 Epstein-Barr virus genome. Nature (London) 310:207-211.
5. Brasier, A. R., J. E. Tate, and J. F. Habener. 1989. Optimized use of the firefly luciferase assay as areporter gene in mamma-lian cell lines. BioTechniques7:1116-1122.
6. Challberg, M. D., and T. J. Kelly. 1989. Animal virus DNA replication. Annu. Rev. Biochem. 58:671-717.
7. Chittenden, T., S. Lupton, and A. J. Levine. 1989. Functional limits of oriP, the Epstein-Barrvirus plasmid origin of replica-tion. J. Virol.63:3016-3025.
8. de Wet, J. R.,K. B. Wood, M. DeLuca, D. R.Helinski, and S. Subramani. 1987. Firefly luciferasegene: structure and expres-sion in mammaliancells. Mol. Cell. Biol. 7:725-737.
9. Frappier, L., and M. O'Donnell. 1991. Overproduction, purifi-cation, and' characterization of EBNA1, the origin binding protein of Epstein-Barr virus. J. Biol. Chem.266:7819-7826. 9a.Frappier, L., and M. O'Donnell. Proc.Natl.Acad. Sci. USA, in
press.
10. Gahn, T. A., and C. L. Schildkraut. 1989. The Epstein-Barr virus origin of plasmid replication, oriP, contains both the initiation and terminationsites of DNA replication. Cell 58:527-535.
11. Hammarskjold,M. L., S. C. Wang, and G. Klein. 1986. High-level expression of the Epstein-Barr virus EBNA1 protein in CV1 cells and human lymphoid cellsusing aSV40late replace-ment vector. Gene 43:41-50.
12. Kadonaga, J. T., and R. Tjian. 1986. Affinity purification of sequence-specific DNAbindingproteins.Proc. Natl.Acad. Sci. USA 83:5889-5893.
13. Kimball, A. S., G. Milman, and T. D. Tullius. 1989. High-resolution footprints of the DNA-binding domain of Epstein-Barr virus nuclear antigen 1. Mol. Cell. Biol.9:2738-2742. 14. Klein-Hitpass, L., M. Schorpp, U. Wagner, and G. U. Ryffel.
1986. An estrogen-responsive element derived from the 5' flanking region oftheXenopusvitellogeninA2 genefunctionsin transfectedhuman cells. Cell46:1053-1061.
15. Knutson, J. C., and D. Yee. 1987. Electroporation: parameters affecting transfer of DNAintomammalian cells.Anal. Biochem. 164:44-52.
16. Lupton, S., and A. J. Levine. 1985. Mappinggeneticelementsof Epstein-Barr virusthat facilitateextrachromosomalpersistence in Epstein-Barr virus-derived plasmids in human cells. Mol. Cell. Biol. 5:2533-2542.
17. Martin, J., and B. Sugden. 1991. Transformationby the onco-genic latent membrane protein correlates with its rapid turn-over, membrane localization, and cytoskeletal association. J. Virol.65:3246-3258.
18. Mertz, J. E., and P. Berg. 1974. Defective simian virus 40 genomes: isolation and growth ofindividual clones. Virology
62:112-124.
19. Milman, G., and E. S.Hwang.1987. Epstein-Barrvirus nuclear antigen forms a complex that binds with high concentration dependenceto asingleDNA-bindingsite. J.Virol. 61:465-471. 20. Nishimura,R., M. J.Raymond,I.Ji,R. V.Rebois,and T. H.Ji. 1986. Photoaffinitylabeling of thegonadotropin receptor with native, asialo, and deglycosylated choriogonadotropin. Proc. Natl.Acad. Sci. USA83:6327-6331.
21. Nordstrom, K. 1990. Control ofplasmid replication-how do DNAiteronssetthereplicationfrequency?Cell 63:1121-1124. 22. Palzkill, T. G., and C. S. Newlon. 1988. A yeast replication
origin consists ofmultiplecopiesofasmall conservedsequence. Cell 53:441-450.
23. Polvino-Bodnar, M.,J. Kiso, and P. A. Schaffer. 1988. Muta-tionalanalysis ofEpstein-Barrvirus nuclearantigen1 (EBNA-1).Nucleic AcidsRes. 16:3415-3435.
24. Ptashne,M.1986. Generegulation byproteinsactingnearbyand at adistance. Nature(London)322:697-701.
25. Rawlins, D.R., G.Milman,S. D.Hayward,and G. S.Hayward.
1985.Sequence-specific DNAbindingof theEpstein-Barrvirus nuclear antigen (EBNA-1) to clustered sites in the
plasmid
maintenanceregion. Cell 42:859-868.
26. Reisman, D., and B. Sugden. 1986. trans Activation of an Epstein-Barr viraltranscriptionalenhancerbytheEpstein-Barr
viralnuclearantigen 1.Mol. Cell. Biol. 6:3838-3846.
27. Reisman, D.,J.Yates,and B.Sugden.1985.Aputative
origin
of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol. Cell. Biol. 5:1822-1832.28. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring
Harbor,
N.Y.29. Schnos, M., K. Zahn, F. R. Blattner, and R. B. Inman. 1989. DNA looping induced by bacteriophage lambda 0
protein:
implications for formation of higher order structures at the lambdaoriginofreplication. Virology 168:370-377.
30. Sternas, L., T. Middleton, and B. Sugden. 1990. The average number of molecules ofEpstein-Barrnuclearantigen 1 percell does not correlate with the average number of
Epstein-Barr
virus(EBV) DNAmolecules percellamongdifferent clones of EBV-immortalized cells. J. Virol. 64:2407-2410.
31. Stillman, B. 1989. Initiation ofeukaryotic DNA
replication
in vitro. Annu. Rev. Cell Biol. 5:197-245.31a.Su, W., T. Middleton, B. Sugden, and H. Echols. Proc. Natl.
Acad. Sci. USA,inpress.
32. Sugden,B.,and N. Warren. 1989. A promoter of
Epstein-Barr
virus thatcanfunctionduringlatent infectioncanbe transacti-vated by EBNA-1, a viral protein required for viral DNA replicationduringlatentinfection.J. Virol. 63:2644 2649. 33. Wysokenski, D. A., and J. L. Yates. 1989.
Multiple
EBNA1-binding sites are required to form an
EBNA1-dependent
en-hancer andtoactivateaminimalreplicativeorigin
within oriP of Epstein-Barr virus.J.Virol.63:2657-2666.34. Yates, J., N. Warren, D. Reisman, and B. Sugden. 1984. A cis-acting element from the Epstein-Barr viral genome that
permits stable replication of recombinant
plasmids
inlatently
infected cells. Proc.Natl. Acad. Sci. USA 81:3806-3810. 35. Yates, J. L., and S. M. Camiolo. 1988. Dissection of DNAreplication and enhancer activation functions of
Epstein-Barr
virus nuclearantigen 1. Cancer Cells 6:197-205.
36. Yates, J. L., and N. Guan. 1991.
Epstein-Barr
virus-derived plasmidsreplicateonlyoncepercellcycle
andarenotamplified
afterentryinto cells. J. Virol. 65:483-488.
37. Yates,J.L.,N.Warren,andB.Sugden. 1985.Stable
replication
ofplasmidsderived from