0022-538X/96/$04.0010
Copyrightq1996, American Society for Microbiology
RNase H Domain of Moloney Murine Leukemia Virus Reverse
Transcriptase Retains Activity but Requires the Polymerase
Domain for Specificity
SHARON J. SCHULTZANDJAMES J. CHAMPOUX*
Department of Microbiology, School of Medicine, University of Washington, Seattle, Washington 98195-7242
Received 12 July 1996/Accepted 30 August 1996
The reverse transcriptase-associated RNase H activity of Moloney murine leukemia virus specifically cleaves within the polypurine tract region of the viral genome to generate the primer for plus-strand DNA synthesis and removes the tRNA primer after minus-strand initiation by preferentially cleaving the RNA one nucleotide before the RNA-DNA junction. Moreover, the enzyme is unable to cleave the extended tRNA substrate at the RNA-DNA junction even at high enzyme concentrations. The RNase H domain of the reverse transcriptase was expressed as a glutathioneS-transferase fusion protein and purified fromEscherichia coliextracts. Following removal of the glutathioneS-transferase portion of the protein, the specificity of the isolated RNase H domain was determined in the plus-strand primer reaction and in the tRNA primer removal reaction. Although the isolated domain lacked specificity in both cases, it was still unable to cleave the tRNA substrate precisely at the RNA-DNA junction. Specificity in both cases could be restored by adding back a truncated form of Moloney murine leukemia virus reverse transcriptase lacking the RNase H domain. These results implicate the poly-merase domain as a specificity determinant for the RNase H activity of reverse transcriptase. The isolated RNase H domain had higher activity in the presence of Mn21than in the presence of Mg21, but neither the RNase H domain alone nor the RNase H domain coupled to the polymerase domain in wild-type protein exhibited the normal cleavage specificities in the presence of the nonphysiological divalent cation.
Upon entering the cytoplasm of a cell, a retrovirus converts its single-stranded plus-sense RNA genome into a linear, dou-ble-stranded DNA in a replication process termed reverse transcription. During reverse transcription, unique sequences at the 59 (U5) and 39 (U3) ends of the viral genome are duplicated through two template jumps that require the direct repeat (R) sequences present at the two ends of the RNA and the tRNA primer binding site (PBS) located immediately downstream of the R-U5 region. Plus-strand DNA synthesis starts from a specific RNA primer created during replication in a region termed the polypurine tract (PPT). The ends of the resulting unintegrated viral DNA contain long terminal repeat sequences that have the structure 59 U3-R-U5 39. The linear viral DNA is then integrated into the host cell chromosome to generate the provirus, from which single-stranded plus-sense RNA genomes as well as viral mRNA are transcribed (8, 19, 51).
Reverse transcription is carried out by the enzyme reverse transcriptase (RT), which possesses an RNase H activity in addition to a DNA polymerase activity. Mutations that elimi-nate either of these activities render a retrovirus noninfectious (50). In addition to using the single-stranded plus-sense RNA and minus-sense DNA strands as templates, the polymerase carries out strand displacement synthesis which is required to complete the viral long terminal repeats (23, 24, 53; for a review, see reference 4). The RNase H activity has three dis-tinct roles during the course of reverse transcription (for a review, see reference 6). First, RNase H degrades the viral genome within the R region to facilitate minus-DNA strand transfer (48) and presumably elsewhere within the genome to
free the minus-strand DNA as a template for plus-strand syn-thesis. Second, RNase H cleaves the PPT in a sequence-de-pendent manner to generate the RNA primer for plus-strand synthesis (13, 15, 25, 27, 32, 35, 42). Third, RNase H specifi-cally removes the minus-strand and plus-strand RNA primers after they have been elongated by the polymerase (7, 15, 16, 25, 28, 33, 35, 38, 44). Interestingly, RNase H of Moloney murine leukemia virus (M-MuLV) and human immunodeficiency virus type 1 (HIV-1) incompletely remove the tRNA primer to leave a ribonucleotide A at the 59end of the minus-strand DNA (16, 33, 38, 44), while that of avian myeloblastosis virus removes the entire tRNA primer (7, 28).
The larger 66-kDa subunit of the heterodimeric HIV-1 RT is a two-domain protein in which the polymerase activity is lo-cated in the amino-terminal two-thirds of the protein and the RNase H activity is located in the remaining one-third (31). In the case of HIV-1, mutations in one domain often affect the activity of the other domain, suggesting that the polymerase and RNase H activities function in an interdependent manner (21, 31). However, an active form of the HIV-1 RNase H has been expressed with an N-terminal histidine tag (45). This form of the RNase H domain is active only in the presence of Mn21but still appears to exhibit the same unique specificity for tRNA primer removal as the wild-type enzyme. While the enzymatic activities of M-MuLV RT are similarly positioned within a single 77-kDa polypeptide chain, the domain bound-ary appears to be more distinct than for HIV-1 (for a review, see reference 30). For example, insertion mutations in M-MuLV RT that destroy either polymerase or RNase H func-tion retain enzymatic activity in the other domain, suggesting that the basic catalytic function of each domain is independent (47). A C-terminal deletion of M-MuLV RT that eliminates the RNase H domain results in a functional polymerase (26), although this domain exhibits reduced processivity (49). Func-tional M-MuLV RNase H has been expressed independent of
* Corresponding author. Mailing address: Department of Microbi-ology, University of Washington, Box 357242, Seattle, WA 98195-7242. Phone: (206) 543-8574. Fax: (206) 543-8297. Electronic mail address: [email protected].
8630
on November 9, 2019 by guest
http://jvi.asm.org/
the polymerase domain, but thus far activity has been mea-sured only by in situ gel analysis in the presence of both Mn21 and Mg21 (2, 47). Thus, for M-MuLV, it remains unclear whether the isolated RNase H domain retains Mg21 -depen-dent activity and exhibits any of the cleavage specificities re-quired for reverse transcription.
To determine whether the specificity of M-MuLV RNase H in the intact RT enzyme is intrinsic to the domain or depen-dent on the presence of the polymerase domain, we have purified and characterized a recombinant form of the RNase H domain. The results indicate that the isolated domain pos-sesses RNase H activity but lacks most of the specificity exhib-ited by the intact enzyme. Mixing experiments using a trun-cated form of M-MuLV RT that lacks the RNase H domain (RNase H2RT) confirmed that the polymerase domain con-fers specificity to the RNase H domain of RT.
MATERIALS AND METHODS
Oligonucleotides and 5* end labeling.For oligonucleotides containing M-MuLV sequences, the sequence positions (39) and polarity relative to the plus-sense viral genome are given after the oligonucleotide sequence. DNA oligonu-cleotide RH-1 (59-GGCCCCCAACTTGGTTAGAGGAGGGTAGAGGTG-39; positions 4595 to 4610 and 4675 to 4691, minus polarity) and DNA oligonucle-otide SS1 (59-GACTTGGTTGAGGCCTCACCAGTCAC-39; converts the ScaI site in the ampicillin resistance gene of pGEM3Zf to a StuI site) were purchased from DNA Express. DNA oligonucleotide M7826 (59-GGGGTCTTTCATTCC CCCCTTTTTCTGG-39; positions 7801 to 7828, minus polarity), DNA oligonu-cleotide 2 (59-TGTGGTCTCGCTGTTCCTTGGG-39; positions 76 to 97, plus polarity), RNA oligonucleotide Mol15R (59-CCGGACGAGCCCCCA-39; posi-tions 146 to 161, minus polarity), and RNA oligonucleotide I (59-AGAAAAAG GGGGGAAUGAAA-39; positions 7803 to 7822, plus polarity) have been de-scribed previously (34, 38). Oligonucleotides were 59end labeled with [g-32P]ATP
(DuPont NEN) as previously described (38).
Recombinant plasmid and phage DNAs.The 1,189-bp SalI-HindIII fragment encoding the carboxy terminus of the polymerase domain, the entire RNase H domain, and the amino terminus of integrase was isolated from p8.2 (40) and inserted into the SalI and HindIII sites of pGEM3Zf (Promega Corp.) to gen-erate pRNaseH-1. By using a double-stranded mutagenesis procedure (10) (Transformer Site-Directed Mutagenesis Kit; Clontech), an in-frame termina-tion codon was placed at the 39end of the sequence coding for the RNase H domain in pRNaseH-1 (2, 47) to produce pRNaseH-2. This was accomplished by using mutagenic oligonucleotide RH-1, which deletes bases 4611 through 4674 in the M-MuLV sequence (39), and selection oligonucleotide SS1. The fragment coding for the RNase H domain was released from pRNaseH-2 by digestion with
EcoRV (which cleaves at the 59end of the sequence encoding the RNase H
domain) and HindIII. After blunt ending, this fragment was inserted at the filled-in BamHI site in the vector pGEX-3X, downstream and in-frame with the sequence coding for the glutathione S-transferase (GST) peptide tag (Pharmacia Biotech), to yield pGEX-RNH. Single-stranded M13mp7/PBS1DNA containing the plus-sense R-U5-PBS sequences (positions 66 to 346 in the M-MuLV ge-nome) was generated as described previously (38).
Enzymes.Recombinant wild-type M-MuLV RT was purchased from U.S. Biochemical Corp. Different lots of the enzyme varied in total protein concen-tration but gave similar responses in our RNase H assays based on the number of units of polymerase activity as given by the manufacturer. Accordingly, the amounts of RT used in the experiments described below are given in terms of polymerase units as designated by the manufacturer. RNase H2RT was pur-chased as Superscript from Life Technologies, Inc. Sequenase version 2.0 was purchased from U.S. Biochemical Corp. Enzymes used for cloning procedures were purchased from New England Biolabs, Inc.
Expression and purification of recombinant proteins.An overnight culture of the TOP10F9strain of Escherichia coli containing pGEX-RNH was diluted 1:10 into 500 ml of LB containing 100mg of ampicillin per ml and 2% glucose and grown for 2 h at 378C. Expression from the pGEX vector was induced by adding isopropylthio-b-D-galactoside (IPTG; Life Technologies) to a final concentration of 0.1 mM, and the culture was grown for 30 to 45 min at 378C. The cultures were cooled on ice for 30 min and centrifuged at 4,3003g for 15 min at 48C. The cell pellet was resuspended in 10 ml of sonication buffer (13phosphate-buffered saline [137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4]
containing phenylmethylsulfonyl fluoride [200 mg/ml], leupeptin [10 mg/ml], aprotinin [1mg/ml], and lysozyme [1mg/ml]), incubated on ice for 5 min, divided into two aliquots, and frozen at2708C. After thawing of a 5-ml aliquot of cells in an ice water bath, an additional 5 ml of sonication buffer was added and the sample was sonicated for 15 s three times. Triton X-100 was added to a final concentration of 1.0%, and the lysate was centrifuged at 10,0003g for 25 min at 48C. The supernatant was mixed with 1 ml of glutathione-Sepharose 4B beads (Pharmacia Biotech) for 60 min at room temperature, and the beads were
pelleted and washed with 13phosphate-buffered saline containing 3% Triton X-100. The GST-RNase H domain fusion protein (GST-RNH) was eluted from the beads, or the RNase H domain protein (RNH) was recovered by digestion of the matrix-bound GST-RNH with 35mg of factor Xa (New England Biolabs) per ml as described previously (1). Recombinant GST-RNH and RNH proteins were stored in 50 mM Tris-Cl (pH 8.0)–100 mM NaCl–1 mM dithiothreitol (DTT)– 50% glycerol at2208C and routinely obtained at concentrations of;600mg/ml. The purified proteins were analyzed by sodium dodecyl sulfate (SDS)-polyacryl-amide gel electrophoresis and visualized by Coomassie blue staining.
Nonspecific RNase H assays.32
P-labeled RNA-DNA hybrids were generated by incubating 4 U of E. coli RNA polymerase (Boehringer Mannheim) with 4mg of circular single-stranded M13mp7/PBS1DNA in a 200-ml reaction containing 50 mM Tris-Cl (pH 8.0), 75 mM KCl, 10 mM MgCl2, 5 mM DTT, 150mM ATP,
150mM CTP, 150mM GTP, 15mM UTP, and 100mCi of [a-32
P]UTP (DuPont NEN) at 378C for 18 h. The reaction was stopped by adding EDTA to 15 mM, and the hybrids were precipitated twice with 2 M ammonium acetate and 2 volumes of ethanol. The hybrids were resuspended in 200ml of TE (10 mM Tris-Cl [pH 8.0], 1 mM EDTA) and heated at 378C for 5 min. After addition of SDS to a final concentration of 0.1%, the products were extracted twice with phenol and twice with chloroform and precipitated with 0.3 M sodium acetate and 2 volumes of ethanol. The hybrids were resuspended in 200ml of TE and routinely had a specific activity of 53106
cpm/mg.
For the in situ gel assay of RNase H activity, the hybrid substrate (;63104
cpm) was polymerized in an SDS–12% polyacrylamide gel. Proteins were elec-trophoresed through the gel and renatured by soaking the gel in several buffer changes of 50 mM Tris-Cl (pH 8.0), 50 mM NaCl, 10 mM MgCl2, 2 mM MnCl2,
and 1 mM DTT for 3 days at room temperature as previously described (47). Cleared bands containing RNase H activity were visualized by autoradiography and are presented as the photographic negative of the autoradiographic image (cleared regions appear as dark bands). For the liquid assay measuring the release of acid-soluble radioactivity, 25-ml reactions containing 2.73105
cpm of the hybrid substrate, 50 mM Tris-Cl (pH 8.0), 1 mM DTT, 100mg of bovine serum albumin (BSA) per ml, and the indicated amount of either MnCl2or
MgCl2were incubated at 378C in the presence of RT, RNH, or GST-RNH. At
indicated times, 5-ml aliquots were removed and added to 95ml of TE containing 200mg of salmon sperm DNA per ml. The undigested substrate was precipitated by the addition of 5% ice-cold trichloroacetic acid, and after 10 min on ice, the samples were centrifuged at 10,0003g for 10 min at 48C. Then 100ml of the supernatant was added to 5 ml of Aquasol (DuPont NEN) and counted in a Beckman LS 3801 scintillation counter. Background was subtracted from each experimental time point, using the averaged value of parallel reactions that contained no enzyme.
Analysis of the 5*ends of minus-sense DNA.RNA oligonucleotide Mol15R was annealed to plus-sense single-stranded DNA containing the R-U5-PBS re-gion of the M-MuLV genome released from M13mp7/PBS1by EcoRI digestion and extended with Sequenase as previously described (38). This substrate was treated with RT, RNH, or GST-RNH in a 10-ml reaction volume containing either 13RT buffer (50 mM Tris-Cl [pH 8.0], 10 mM MgCl2, 0.8 mM DTT) or
50 mM Tris-Cl (pH 8.0), 1 mM DTT, 100mg of BSA per ml, and the indicated amount of MgCl2or MnCl2. Reactions were terminated by addition of EDTA to
10 mM, split in two, and either precipitated directly with 0.3 M sodium acetate and 2 volumes of ethanol or first treated with 0.3 M NaOH for 20 min at 658C (to hydrolyze the RNA), neutralized, and then ethanol precipitated. Primer extensions were performed with RNase H2RT essentially as described previ-ously (33). For size markers, sequencing ladders were prepared as previprevi-ously described (38). Samples were analyzed in 8% polyacrylamide gels containing 8.3 M urea (Sequagel; National Diagnostics).
Assay for PBS primer removal.59-end-labeled RNA oligonucleotide Mol15R was annealed to and extended on plus-sense single-stranded M13mp7/PBS1 DNA containing the R-U5-PBS region of the M-MuLV genome as described previously (33, 38). The resulting hybrid substrate was treated in reactions with RT, RNH, or GST-RNH as described above. Aliquots removed from reactions at the indicated times were added to an excess volume of formamide stop buffer (80% deionized formamide, 1 mM EDTA, 0.2% bromphenol blue, 0.2% xylene cyanol) and analyzed in a 20% polyacrylamide gel containing 8.3 M urea.
Assay for cleavage at the PPT.A32
P-labeled RNA-DNA hybrid substrate containing the PPT was prepared by heating 7 pmol of 59-end-labeled RNA oligonucleotide I with 21 pmol of DNA oligonucleotide M7826 in 25 mM Tris-Cl (pH 8.0)–200 mM NaCl at 808C and slowly cooling to room temperature. Hybrids were precipitated with ethanol and resuspended in TE. Hybrids were treated with RT, RNH, or GST-RNH as described above. Samples were mixed with an equal volume of formamide stop buffer and analyzed in a 20% polyacrylamide gel containing 8.3 M urea.
RESULTS
Expression and purification of the M-MuLV RNase H do-main. Previous studies have established that the minimum RNase H domain that is active begins at amino acid 500 in M-MuLV RT (2, 47). We generated a pGEX-3X recombinant
on November 9, 2019 by guest
http://jvi.asm.org/
clone that expresses GST joined to the RNase H domain be-ginning at amino acid 498 in M-MuLV RT and ending with the C terminus for RT 174 amino acids downstream. This con-struct was predicted to produce a 45.8-kDa GST-RNH fusion protein. A comparison of the total cellular proteins expressed in uninduced and induced cultures of E. coli containing this vector revealed the expression of a protein of;43 kDa after induction with IPTG (Fig. 1A; compare lanes 3 and 4). This expression pattern was not observed upon induction of a cul-ture containing the control plasmid which expressed only the GST protein (data not shown).
The recombinant protein was purified from induced cell lysates by using affinity chromatography with glutathione-Sepharose beads under nondenaturing conditions (41). The purified protein preparation consisted almost exclusively of the 43-kDa GST-RNH protein, but a minor protein with a mobility of;38 kDa was also recovered (Fig. 1A, lane 5). Factor Xa digestion of GST-RNH coupled to glutathione-Sepharose beads released a;24-kDa protein representing the RNH por-tion of the fusion protein that was free of GST protein (Fig. 1A, lanes 6 and 7). The recombinant RNH protein contains
two extra amino-terminal amino acids (Gly and Ile) that are present between the factor Xa cleavage site and the start of the M-MuLV RNase H domain in GST-RNH and has a predicted size of 19.8 kDa. Thus, the observed mobility of the purified RNH protein in an SDS-polyacrylamide gel was slightly slower than that predicted from amino acid content.
To test whether the purified recombinant RNH and GST-RNH proteins exhibited RNase H activity, we used an in situ gel assay (47). The proteins were electrophoresed in a dena-turing SDS-polyacrylamide gel containing a uniform concen-tration of 32P-labeled RNA-DNA hybrid substrate and then renatured through several buffer changes (see Materials and Methods). After renaturation, localized degradation of the substrate in the vicinity of any active RNase H protein band was detected as a cleared area on the resulting autoradiogram or as a dark band in the negative image shown in Fig. 1B. Lysates from uninduced and induced cell cultures contained several proteins with RNase H activity (Fig. 1B, lanes 1 and 2), but the 43-kDa GST-RNH protein was present only in the induced cell lysate (Fig. 1B, lane 2). The two smaller promi-nent protein bands in the uninduced sample (Fig. 1B, lane 1) corresponded to E. coli RNase H species. Proteins smaller than GST-RNH but larger than the bacterial forms were evi-dent exclusively in induced cell lysates (Fig. 1B, lane 2) and likely represented proteolytic fragments of GST-RNH that retained RNase H activity. After purification with glutathione-Sepharose beads, GST-RNH retained activity and increasing amounts of this preparation failed to reveal any contaminating
E. coli RNase H activities (Fig. 1B, lanes 3 to 5). The RNH
protein generated by digestion with factor Xa exhibited signif-icantly higher levels of RNase H activity than GST-RNH, even after correction for the relative molar amounts of the two proteins present in the gel (Fig. 1B; compare lanes 6 to 8 with lanes 3 to 5). Wild-type M-MuLV RT also displayed the ex-pected RNase H activity without any contaminating activities (Fig. 1B, lanes 9 and 10). When a single-stranded32P-labeled RNA was used as the substrate in an otherwise identical assay, the recombinant proteins as well as wild-type M-MuLV RT lacked any detectable single-stranded RNase activity (data not shown). These analyses demonstrated that the purified recom-binant RNH and GST-RNH proteins retained RNase H activ-ity and were free of any contaminating RNase activities.
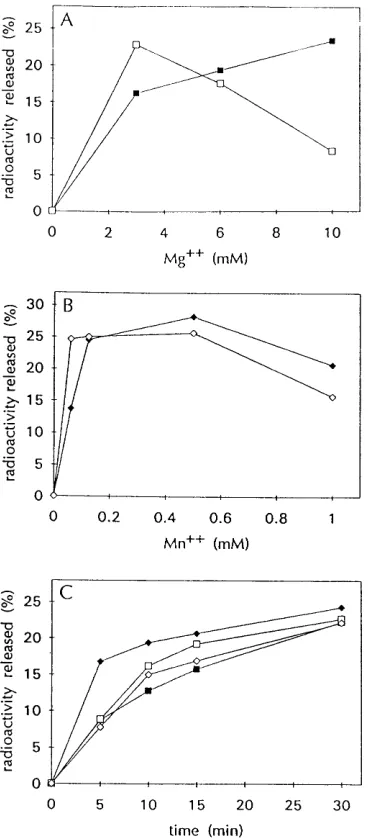
Optimum reaction conditions for comparing RNase H ac-tivities of RT, RNH, and GST-RNH.To establish the condi-tions for maximal activities of recombinant RNH, GST-RNH, and RT proteins in the same reaction conditions, we used a liquid assay that measured nonspecific RNase H activity by the release of acid-soluble radioactivity from a32P-labeled RNA-DNA hybrid substrate (45). Initially we compared the divalent cation requirements of RT and RNH. As shown in Fig. 2A, when the Mg21 concentration was varied between 0 and 10 mM, the RNase H activity of RT was highest at 10 mM Mg21 while that of RNH was optimal at 3 mM Mg21. Since increas-ing Mg21concentrations had opposite effects on the RNase H activities of RT and RNH, 6 mM Mg21 was chosen as the compromise for subsequent reactions that compared the two enzymes. Much lower concentrations of Mn21were required to achieve similar levels of RNase H activity for RT and RNH (Fig. 2B). For both enzymes, 0.5 mM Mn21 generated the highest levels of activity.
[image:3.612.105.248.71.329.2]Using either 6 mM Mg21or 0.5 mM Mn21, we established the relative amounts of RT and RNH proteins required to achieve the same level of nonspecific RNase H activity. Rep-resentative data from these kinetic analyses are presented in Fig. 2C. Twelve and a half units of RT had approximately the same level of activity as 600 ng of RNH in 6 mM Mg21, while FIG. 1. Purification and RNase H activities of recombinant RNH and
GST-RNH proteins. Proteins were subjected to electrophoresis in SDS–12% poly-acrylamide gels and visualized by staining with Coomassie blue in panel A or assayed in situ and visualized by autoradiography in panel B. (A) Total cell lysates were prepared from uninduced (U; lane 3) or induced (I; lane 4) bacterial cultures expressing pGEX-RNH by resuspending pelleted cells in 23sample buffer (100 mM Tris-HCl [pH 6.8], 4% SDS, 20% glycerol, 2-mercaptoethanol, 0.02% bromphenol blue) and boiling. Lane 5 contained 1.5 g of purified GST-RNH prepared by affinity chromatography with glutathione-Sepharose beads. Lane 6 contained 1.5mg of purified RNH recovered after factor Xa digestion. Lane 7 contained 4mg of purified GST. Lane 1 shows protein size standards with sizes of 190, 125, 88, 65, 56, 38, and 33.5 kDa from top to bottom, and lane 2 shows protein size standards which are indicated at the right. (B) Lanes 1 and 2 contained the uninduced total cell lysate and the induced total cell lysate, re-spectively. Lanes 3 to 5 contained 1.5, 3, and 6 mg of purified GST-RNH, respectively. Lanes 6 to 8 contained 1.5, 3, and 6mg of purified RNH, respec-tively. Lanes 9 and 10 contained 200 U (0.84mg) and 400 U (1.7mg) of RT, respectively. Protein size standards are indicated at the left. The in situ gel assay of RNase H activity is shown as the negative image such that areas of enzyme activity are black and areas without activity remain white.
on November 9, 2019 by guest
http://jvi.asm.org/
10 U of RT was comparable to 4 ng of RNH in 0.5 mM Mn21. In a series of identical assays with GST-RNH, the RNase H activity of the fusion protein paralleled that presented above for RNH except that approximately twice as much GST-RNH protein was required (data not shown), consistent with its greater molecular weight.
Other parameters affecting the activity of RNase H were similarly examined to determine the optimal conditions for comparing the activities of RT, RNH, and GST-RNH. Previ-ous studies have shown that recombinant retroviral RNases H typically have maximal activity between pH 6.8 and 8.8 (12, 45). In a pH titration over this range, RT and RNH exhibited optimal activities at pH 8 whereas that of GST-RNH was
optimal at pH 6.8 but not greatly reduced at pH 8.0 (data not shown). KCl was omitted from these reactions since the RNase H activity of RT is maximal in the absence of KCl (5, 14) and the salt concentration does not affect the specificity of PBS primer removal for M-MuLV RT (38). During these studies, we found that the addition of 100mg of BSA per ml to the reaction mixtures significantly increased the amount of activity observed in the acid solubility assay without having any effect on the different specificity assays used for RNase H. When we substituted 32P-labeled single-stranded RNA for the 32 P-la-beled RNA-DNA hybrid in the nonspecific assay, the recom-binant RNH and GST-RNH preparations contained negligible levels of RNase activity (data not shown).
Based on these analyses, the reactions for assaying the RNase H activity of RT, RNH, and GST-RNH contained 50 mM Tris-HCl (pH 8.0), 1 mM DTT, 100mg of BSA per ml, and either 6 mM MgCl2or 0.5 mM MnCl2. The relative specific activities of RT, RNH, and GST-RNH under these reaction conditions are summarized in Table 1.
[image:4.612.87.271.71.490.2]Specificity of tRNA primer removal by M-MuLV RNase H domain. We previously showed that the RNase H activity of M-MuLV RT removes the minus-strand tRNA primer by pref-erentially cleaving one nucleotide away from the RNA-DNA junction (38). To address whether the isolated RNase H do-main of M-MuLV retained this specificity when separated from the polymerase domain of RT, we initially examined whether the recombinant RNH exhibited the same cleavage preference as RT, using model substrates in vitro. RNA oligo-nucleotide Mol15R was extended on single-stranded plus-sense DNA containing the U5-R-PBS sequences of M-MuLV to generate a DNA duplex which includes the PBS RNA primer and represents the substrate for RNase H cleavage prior to the second jump of reverse transcription (19). This substrate was incubated with RT, RNH, or GST-RNH in the absence of deoxynucleoside triphosphates (dNTPs), and the RNase H-specific cleavages of the RNA primer were mapped by primer extension analysis using a 59-end-labeled oligonucle-otide that was complementary to the minus-strand DNA. In the absence of enzyme, the primer extension product extended to the 59 end of the RNA primer (Fig. 3, lane 2, arrow a), whereas prior treatment of the substrate with alkali completely removed the RNA primer and exposed the 59 end of the minus-strand DNA (Fig. 3, lane 1, arrow c). As we have pre-viously shown (38), incubating this substrate with 100 U of RT in a reaction buffer containing 10 mM Mg21generated a major product that extended one base beyond the 59end of minus-strand DNA (Fig. 3, lane 10, arrow b). This base was removed by alkali treatment (Fig. 3, lane 9, arrow c), confirming that RT incompletely removed the PBS primer to leave a single ribo-nucleotide A on the end of the DNA strand. This specificity was not affected by reducing the Mg21concentration from 10 to 6 mM (Fig. 3, lane 12). When RT was diluted to 10 U, matching its RNase H activity in the nonspecific assay (see Fig. 2) to that of RNH, the primary product still retained the ribonucleotide A, but additional cleavages 59to this site were also apparent (Fig. 3, lane 4). Treating the model substrate FIG. 2. Analysis of optimal reaction conditions to compare RNase H
activ-ities of RT and RNH. RNase H activity is presented as the percentage of the total radioactivity rendered acid soluble after trichloroacetic acid precipitation as a function of Mg21concentration (A), Mn21concentration (B), or time (C). In panels A and B, assays were performed for 15 min with the indicated amounts of Mg21or Mn21, using 12.5 U of RT (filled boxes), 20 U of RT (filled diamonds), 600 ng of RNH (open boxes), or 60 ng of RNH (open diamonds). In panel C, assays were carried out in the presence of 6 mM Mg21(boxes) or 0.5 mM Mn21 (diamonds), using 12.5 U of RT (filled boxes), 8 U of RT (filled diamonds), 600 ng of RNH (open boxes), or 4 ng of RNH (open diamonds).
TABLE 1. RNase H activities of RT, RNH, and GST-RNH
Protein
Relative sp act
6 mM Mg21 0.5 mM Mn21
RT 43 54
RNH 1 150
GST-RNH 1 91
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.315.556.83.146.2]with either RNH or GST-RNH did not reveal a single pre-ferred cleavage site but instead produced a number of exten-sion products in approximately equal amounts, suggesting that the preference for cleavage between the ribonucleotide A and the penultimate ribonucleotide C exhibited by RT was absent (Fig. 3, lanes 6 and 8). However, despite the absence of a cleavage preference, no extension product corresponding to precise cleavage at the RNA-DNA junction was observed for RNH or GST-RNH (Fig. 3, lanes 6 and 8), even when the substrate was treated with the enzymes for as long as 120 min (data not shown).
The same in vitro model substrate was also used to examine whether Mn21affected the cleavage specificity of the various forms of M-MuLV RNase H. Unlike the cleavage pattern observed with Mg21, both higher and lower amounts of RT in the presence of Mn21generated essentially identical results in which the substrate was cleaved at several sites (Fig. 4, lanes 3 and 4). While Mn21relaxed the specificity for cleavage some-what, the predominant product still contained the ribonucle-otide A (arrow b) and there was very little cleavage at the RNA-DNA junction (arrow c). In the presence of Mn21, the cleavage patterns for both RNH and GST-RNH resembled that of RT except that there was no preference for cleavage one nucleotide away from the RNA-DNA junction (Fig. 4, lanes 5 and 6).
Fate of the RNA primer.To address the fate of the RNA primer in the RNase H cleavage assay, 59-end-labeled RNA oligonucleotide Mol15R was extended on a single-stranded DNA containing the U5-R-PBS sequences and used as a sub-strate for RT or RNH. As shown in Fig. 5A, the extended substrate migrated much slower than the 15-mer RNA oligo-nucleotide (compare lanes 1 and 6 with lane 11). When this
[image:5.612.341.525.71.227.2]substrate was incubated with RT for as little as 1 min in 6 mM Mg21, the major product migrated as a 14-mer (Fig. 5A, lane 2), consistent with the initial preferred cleavage by RT occur-ring between the ribonucleotide A and penultimate ribonucle-otide C. With increasing time, most of the extended substrate was cleaved by RT and the final cleavage products were pre-dominantly fragments 7 and 8 nucleotides in length (Fig. 5A, lanes 4 and 5). Unlike RT, RNH did not produce any of the 14-mer product, but similar to RT, cleavage products 7 and 8 nucleotides in length were generated (Fig. 5A, lanes 7 to 9). After 27 min of incubation with RNH, most of the extended substrate was digested and a spectrum of cleavage products ranging from 4 to 14 nucleotides in length was evident (Fig. 5A, FIG. 3. Analysis of the 59end of minus-strand DNA at the U5-PBS junction
[image:5.612.62.293.72.220.2]after primer removal in the presence of Mg21. RNA oligonucleotide Mol15R was annealed to a linear single-stranded DNA containing the plus-sense R-U5-PBS sequence of M-MuLV and extended with Sequenase to generate a DNA-DNA duplex that models the structure of minus-strand DNA-DNA prior to primer removal. This substrate was left untreated (control; lanes 1 and 2) or treated with 10 U of RT (lanes 3 and 4), 600 ng of RNH (lanes 5 and 6), 1,200 ng of GST-RNH (lanes 7 and 8), or 100 U of RT (lanes 9 to 12) for 15 min in the presence of 6 mM (lanes 1 to 8, 11, and 12) or 10 mM (lanes 9 and 10) Mg21. One-half of each sample was treated with alkali (lanes 1, 3, 5, 7, 9, and 11), and the 59ends of minus-strand DNA for both the treated and untreated samples were mapped by primer extension using 59-end-labeled oligonucleotide 2 and Superscript. Samples were analyzed in a 20% polyacrylamide–8.3 M urea gel adjacent to a dideoxy sequencing gel ladder generated by using 59-end-labeled oligonucleotide 2 and M13mp7/PBS1DNA (lanes T, G, C, and A). The se-quence of the RNA oligonucleotide is indicated at the left. For the extension products, arrow a denotes the position corresponding to the 59end of the RNA primer, arrow b denotes the 59end of minus-strand DNA containing an extra ribonucleotide A, and arrow c denotes the 59end of minus-strand DNA.
FIG. 4. 59-end analysis of minus-strand DNA after primer removal in the presence of Mn21. Substrates were analyzed in 0.5 mM Mn21for 15 min as described in the legend to Fig. 3 and left untreated (lanes 2 to 6) or treated with alkali (lane 1) prior to primer extension analysis. Lanes 1 and 2, no enzyme (control); lane 3, 200 U of RT; lane 4, 10 U of RT; lane 5, 4 ng of RNH; lane 6, 15 ng of GST-RNH. The designations for the arrows and the sequence of the RNA oligonucleotide are as indicated for Fig. 3.
FIG. 5. Time course of PBS primer cleavage in the presence of Mg21or Mn21. After extension of 59-end-labeled RNA oligonucleotide Mol15R on M13mp7/PBS1, the resulting substrate was gel purified and used to assay RNase H cleavages on the PBS primer. Samples were analyzed by electrophoresis in a 20% polyacrylamide–8.3 M urea gel. (A) Cleavage in the presence of 6 mM Mg21. The substrate (untreated in lanes 1 and 6) was incubated with 10 U of RT (lanes 2 to 5) or with 600 ng of RNH (lanes 7 to 10) for the indicated times. The 15-mer RNA oligonucleotide Mol15R is shown in lane 11. (B) Cleavage in the presence of 0.5 mM Mn21. The substrate (untreated in lanes 1 and 5) was incubated with 2.5 U of RT (lanes 2 to 4) or 4 ng of RNH (lanes 6 to 8) for the indicated times. The 15-mer RNA oligonucleotide is shown in lane 9. Nucleotide lengths are indicated to the right of each panel.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.318.551.459.626.2]lane 10). Consistent with the results from the primer extension analyses above, a 15-mer corresponding to cleavage exactly at the RNA-DNA junction was not detected at any time for either RT or RNH.
In the presence of Mn21, RT and RNH demonstrated sim-ilar cleavage patterns for primer removal. The primer was cleaved as early as 0.3 min, although RT generated a higher proportion of 14-mer relative to 7-mer than RNH (Fig. 5B; compare lanes 2 and 6). With increasing lengths of time, both enzymes continued cleaving the RNA primer to yield primarily 7-mers (Fig. 5B, lanes 3, 4, 7, and 8). When this substrate was treated with GST-RNH in the presence of Mg21or Mn21, the cleavage products were essentially identical to those observed for RNH (data not shown).
Specificity of PPT cleavage by M-MuLV RNase H domain.
During reverse transcription, RNase H specifically cleaves within the PPT RNA to generate the plus-strand DNA primer. To examine whether the polymerase domain of RT is impor-tant for this cleavage specificity, we again used a model sub-strate in vitro. 59-end-labeled RNA oligonucleotide I, contain-ing 13 nucleotides 59and 7 nucleotides 39of the plus-strand primer origin, was annealed to a 28-mer DNA oligonucleotide that contained all of the sequences complementary to RNA oligonucleotide I plus an additional six unpaired bases extend-ing 59and two unpaired bases extending 39of oligonucleotide I. This duplex substrate was treated with RT or RNH in the absence of dNTPs, and the resulting products were analyzed by electrophoresis in a denaturing gel. Incubation of the substrate with RT resulted in several cleavages at the earliest time point. The major cleavage products were 13-mers, representing age at the plus-strand origin, and 15-mers resulting from cleav-age two bases downstream from the origin (Fig. 6, lane 2). While RT continued to cleave the unprocessed substrate throughout the time course of the reaction, virtually no frag-ments of 12 bases or smaller reflecting cleavages internal to the PPT were observed (Fig. 6, lanes 3 to 5). In contrast, RNH did not cleave the substrate as rapidly as RT, but the observed cleavages were much more extensive and occurred both
up-stream and downup-stream of the plus-strand origin site (Fig. 6, lanes 6 to 9). Substituting Mn21 for Mg21 as the divalent cation in the PPT cleavage assay relaxed the specificity of RT, resulting in similar product distributions for both RT and RNH. For both enzymes, cleavages at the PPT origin and downstream were preferred early in the time course (Fig. 6, lanes 10 and 14). With longer times, the RNA oligonucleotide was progressively cleaved within the PPT to produce smaller fragments (Fig. 6, lanes 11 to 13 and 15 to 17). When GST-RNH was tested in this assay in the presence of Mg21 and Mn21, results were essentially identical to those observed for RNH (data not shown).
Reconstituting RNase H specificity by combining RNase H2 RT with RNase H domain. The naturally occurring HIV-1 RNase H domain (p15) is inactive alone but exhibits activity when combined with the polymerase domain (p51) in vitro (22). Although the isolated M-MuLV RNase H domain de-scribed in this study retains RNase H activity, it is unable to specifically remove the PBS primer or generate the PPT primer. As for the HIV-1 experiments, we examined whether RNase H specificity requires that the polymerase domain be covalently joined to the RNase H domain, or whether the polymerase domain can function in trans. For the polymerase domain, we used recombinant M-MuLV RNase H2RT (Su-perscript), which contains the first 495 residues of RT (26) and therefore terminates two amino acids before the beginning of our recombinant RNH.
[image:6.612.79.280.68.258.2]Using the model substrate for tRNA primer removal with the 59end of the RNA labeled, we found that a twofold molar excess of RNase H2RT over RNH was able to partially re-store the cleavage preference of RNH to the site one ribonu-cleotide away from the RNA-DNA junction (Fig. 7). This effect was particularly pronounced at the 3-min time point, FIG. 6. Time course of PPT cleavage in the presence of Mg21or Mn21.
59-end-labeled RNA oligonucleotide I was annealed to DNA oligonucleotide M7826 to generate a duplex containing the PPT and used as a substrate to assay RNase H cleavages in the presence of 6 mM Mg21(lanes 2 to 9) or 0.5 mM Mn21(lanes 10 to 17). The substrate was incubated with 10 U of RT (lanes 2 to 5 and 10 to 13), 600 ng of RNH (lanes 6 to 9), or 4 ng of RNH (14 to 17) for the indicated times. The untreated substrate is shown in lane 1, and nucleotide lengths are indicated at the right.
FIG. 7. Combining RNase H2RT with RNH during PBS primer removal. The hybrid substrate containing 59-end-labeled RNA oligonucleotide Mol15R was prepared and analyzed as described for Fig. 5. All reactions were carried out under standard conditions with 6 mM Mg21. The substrate was left untreated (lane 1) or incubated with 10 U of RT (lanes 2 and 3), 600 ng of RNH (lanes 4 and 5), 600 ng of RNH plus 3mg of RNase H2RT (lanes 6 and 7), or 3mg of RNase H2RT alone (lane 8) for the indicated times. The 15-mer RNA oligo-nucleotide Mol15R is shown in lane 9. The mobilities of the 14- and 15-mer RNA oligonucleotides are indicated on the right.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.354.503.72.293.2]where the cleavage pattern and the production of the 14-mer product by the mixture closely resembled the pattern for RT (Fig. 7; compare lanes 2, 4, and 6). By 30 min, more extensive internal cleavage by the mixture was evident, but the 14-mer product remained and, as before, was completely absent in the RNH alone control (Fig. 7, lanes 5 and 7). A control reaction containing a vast excess of RNase H2RT over RT (95-fold molar excess) showed that the truncated RT had no effect on the cleavage pattern of RT (data not shown).
We also determined the effects of adding RNase H2RT to RNH reactions containing the model duplex oligonucleotide substrate for assaying PPT primer cleavage. Incubating this substrate with RT and a vast excess of RNase H2RT resulted in a slight reduction in the kinetics of cleaving the PPT, but the overall pattern of cleavage remained similar to that observed for RT alone (Fig. 8A; compare lanes 6 to 9 with lanes 2 to 5; Fig. 8B, lanes 2 to 5). In contrast, adding a twofold molar excess of RNase H2 RT to RNH diminished the level of nonspecific cleavages upstream of the plus-strand origin site while increasing cleavages downstream of the plus-strand ori-gin (Fig. 8A; compare lanes 15 to 18 with lanes 11 to 14). In the absence of RNase H2RT, RNH generated proportionally very little of either the 13-mer plus-strand primer or the 15-mer cleavage product, but in the presence of increasing amounts of the truncated RT, the cleavages at the plus-strand origin and downstream of the origin were enhanced and cleavages within the PPT region were nearly eliminated (Fig. 8B, lanes 6 to 9).
DISCUSSION
Two features characterize removal of the tRNA primer by M-MuLV RT. First, like the HIV-1 RNase H activity, the enzyme preferentially cleaves the RNA one nucleotide away from the RNA-DNA junction to leave a ribonucleotide A on the 59end of the minus-strand DNA (16, 33, 38, 44). Second, based on experiments in vitro, the M-MuLV RT appears un-able to remove the RNA by cleavage precisely at the RNA-DNA junction (38), despite the fact that cleavage at the junc-tion occurs efficiently for the removal of the PPT plus-strand primer (34–36). Since we show here that the RNase H domain
alone is relatively nonspecific and shows no preference for cleavage one base away from the junction, the polymerase domain is likely responsible for conferring this level of speci-ficity (see below). In contrast, the inability to cleave at the tRNA-DNA junction appears to be intrinsic to the RNase H activity. The isolated HIV-1 RNase H domain (p15) exhibited no activity (22, 45), but the addition of an N-terminal histidine tag produced an active form of the protein that was reported to retain the preference for cleaving the tRNA substrate one nucleotide before the RNA-DNA junction (45). Based on this observation, we wondered whether any N-terminal addition might similarly restore specificity to the isolated M-MuLV RNase H domain. However, we found that the GST-RNH lacked specificity in all of the cleavage assays. An alternative explanation for the apparent specificity of the histidine-tagged HIV-1 RNase H domain is that the enzyme levels used in the analysis were sufficiently high that even at the earliest time point examined, most of the substrate had been cleaved (45). Under these conditions, the production of the final cleavage product, which reflects the inability of the HIV-1 RNase H to cleave at the RNA-DNA junction, might have masked an al-teration in the cleavage preference of the enzyme.
[image:7.612.150.472.71.249.2]The generation and preservation of the plus-strand primer by RNase H depend on both specific cleavage at the end of six G residues near the 39end of the PPT and the relative resis-tance of the upstream region to further cleavage by RNase H (13, 34, 35). Both of these properties are absent in the isolated M-MuLV RNase H domain. Many studies indicate that the RNase H active sites of RTs are spatially located approxi-mately 18 template bases away from the primer terminus in actively synthesizing complexes (11, 14, 17, 20, 29, 37). We previously demonstrated that contacts between PPT hybrids and M-MuLV RT as far as 7 bp upstream of the cleavage site are critical for correctly positioning RNase H for the cleavage reaction that creates the plus-strand primer (36). Thus, it ap-pears likely that contacts between the PPT hybrid substrate and the primer-template binding cleft within the polymerase domain are crucial for directing specific cleavage by RNase H at the plus-strand origin. Presumably, similar contacts account for the cleavage preference described above for tRNA primer FIG. 8. Combining RNase H2RT with RNH during PPT cleavage. A hybrid duplex containing the PPT was prepared and analyzed as described for Fig. 6. All reactions were carried out under standard conditions with 6 mM Mg21. (A) The substrate was left untreated (lanes 1 and 10) or treated with 10 U of RT (lanes 2 to 5), 10 U of RT plus 3mg of RNase H2RT (lanes 6 to 9), 600 ng of RNH (lanes 11 to 14), or 600 ng of RNH plus 3mg of RNase H2RT (lanes 15 to 18) for the indicated times. (B) The substrate was incubated for 15 min with 10 U of RT (lanes 2 to 5) or 600 ng of RNH (lanes 6 to 9) either alone (lanes 2 and 6) or in the presence of 0.2mg (lanes 3 and 7), 0.75mg (lanes 4 and 8), or 3mg (lanes 5 and 9) of RNase H2RT. The substrate was also incubated with 3mg of RNase H2RT alone (lane 1).
on November 9, 2019 by guest
http://jvi.asm.org/
removal. The paucity of cleavages throughout the PPT by RT cannot be explained simply by an unusual structure or se-quence that is resistant to the viral RNase H, since we observed fairly uniform cleavage along the entire length of the PPT by the isolated RNase H domain. Instead, the resistance of the upstream sequences within the PPT to degradation likely de-rives from the preferred binding arrangement of the enzyme described above. In addition, once the preferred cleavage at the plus-strand origin has occurred, polymerase binding at the newly created RNA primer terminus may occlude the up-stream region from further cleavage by RNase H.
Metal ions appear critical for both the polymerization func-tion and the RNase H activity of RT. The crystal structure of the finger and palm domains of M-MuLV RT (residues 10 to 278) reveals a single divalent metal ion binding site involving the polymerase active site aspartate residues at positions 224 and 225 (18). This metal ion is likely involved in binding of the primer and in the catalysis of chain elongation (46). The HIV-1 RNase H domain binds either one or possibly two divalent metal ions that are essential for RNA hydrolysis (9, 54). It has been shown previously that both the polymerase activity and the RNase H activity of M-MuLV RT exhibit a higher activity in Mn21 than in Mg21 (3, 52) (Table 1). For the isolated RNase H domain itself, we show here that the differences are even more dramatic, with normalized activities in excess of 100-fold greater with 0.5 mM Mn21than with 6 mM Mg21 (Table 1). Because the RNase H activity of wild-type RT loses specificity in the presence of Mn21and the isolated RNase H domain lacks specificity with either cation, it seems likely that Mg21bound to the polymerase active site plays an important role in determining the specificity of RNase H cleavage. Al-though we consider it unlikely, we cannot rule out the possi-bility that the loss of specificity observed in the presence of Mn21reflects an altered structure of the DNA-RNA hybrid substrate.
We offer the following hypothesis as an attempt to unify the various observations concerning the roles of the polymerase domain and the bound divalent metal ion on the activity and specificity of the RNase H domain. With Mg21bound to the polymerase active site, the polymerase domain actively partic-ipates in the binding of an RNA-DNA hybrid and is largely responsible for conferring on the RNase H activity its prefer-ence for cleaving at particular sites within the tRNA or the PPT. It is noteworthy that Mg21is the metal ion present in vivo during the process of viral reverse transcription. In contrast, with Mn21in the polymerase active site, we suppose that the polymerase domain only weakly contacts the RNA-DNA hy-brid and consequently the RNase H domain exhibits a loss of specificity. This loss is comparable to that observed for the isolated RNase H domain alone with either cation. Further-more, if the rate-limited step in the RNase H reaction is dis-sociation of the enzyme from the substrate, then one can ac-count for the cleavage kinetics of the different forms of RNase H on a nonspecific substrate from the foregoing consider-ations. The weaker interaction of the polymerase domain in the presence of Mn21than in the presence of Mg21would be expected to lead to an increase in the off rate for RT and could account for the observed increase in the rate of the reaction. Similarly, the absence of the polymerase domain altogether should lead to an even higher rate of dissociation, accounting for the much faster kinetics of cleavage of the isolated RNase H domain irrespective of the divalent cation.
By mixing the truncated form of M-MuLV RT missing the RNase H domain with purified RNH, we found that we can partially reconstitute the specificity for cleaving both the tRNA and at the plus-strand origin within the PPT. Therefore, the
ability of the polymerase domain to influence the specificity of the RNase H domain does not require that the two domains be part of the same polypeptide chain. Similar results have been reported for the two domains of HIV-1 RT (43). There are two possible explanations for the reconstitution of RNase H spec-ificity by mixing RNase H2RT and RNH. First, the two sep-arately folded domains of the protein may interact directly and reconstitute a functional protein that possesses the specificity of the intact enzyme. Alternatively, the polymerase domain may bind initially with the RNA-DNA hybrid substrate and thereby position the binding of the RNase H domain to deter-mine its cleavage specificity. Further experiments are required to distinguish between these possibilities.
Interestingly, two different polymerase-defective mutants of M-MuLV RT have been shown to complement an RNase H mutant in phenotypically mixed virions in vivo (50). Presum-ably the polymerase and RNase H activities on separate polypeptides can cooperate to accomplish the process of re-verse transcription. Based on our findings in this study, we would predict that neither of these mutations so drastically alters the overall structure of the polymerase domain that it is unable to provide the proper specificity determinants for RNase H in the plus-strand priming and primer removal reac-tions.
ACKNOWLEDGMENTS
This work was supported by grant R37 CA51605 from the National Institutes of Health.
We thank Samuel Whiting and Lance Stewart for helpful comments and suggestions during the course of these experiments.
REFERENCES
1. Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.).1996. Current protocols in molecular biology. John Wiley & Sons, Inc., New York.
2. Blain, S. W., and S. P. Goff. 1993. Nuclease activities of Moloney murine leukemia virus reverse transcriptase. J. Biol. Chem. 268:23585–23592. 3. Blain, S. W., and S. P. Goff. 1996. Differential effects of Moloney murine
leukemia virus reverse transcriptase mutations on RNase H activity in Mg21 and Mn21. J. Biol. Chem. 271:1448–1454.
4. Boone, L. R., and A. M. Skalka. 1993. Strand displacement synthesis by reverse transcriptase, p. 119–133. In A. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
5. Brewer, L. C., and R. D. Wells. 1974. Mechanistic independence of avian myeloblastosis virus DNA polymerase and ribonuclease H. J. Virol. 14:1494– 1502.
6. Champoux, J. J. 1993. Roles of ribonuclease H in reverse transcription, p. 103–117. In A. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
7. Champoux, J. J., E. Gilboa, and D. Baltimore. 1984. Mechanism of RNA primer removal by the RNase H activity of avian myeloblastosis virus reverse transcriptase. J. Virol. 49:686–691.
8. Coffin, J. M. 1990. Retroviridae and their replication, p. 1437–1500. In B. N. Fields, D. M. Knipe, R. M. Chanock, M. S. Hirsch, J. L. Melnick, T. P. Monath, and B. R. Roizman (ed.), Virology, 2nd ed. Raven Press, Ltd., New York.
9. Davies, J. F., II, Z. Hostomska, Z. Hostomsky, S. R. Jordan, and D. A. Matthews.1991. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science 252:88–95.
10. Deng, W. P., and J. A. Nickoloff. 1992. Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal. Biochem. 200:81–88. 11. DeStefano, J. J., L. M. Mallaber, P. J. Fay, and R. A. Bambera. 1993.
Determinants of the RNase H cleavage specificity of human immunodefi-ciency virus reverse transcriptase. Nucleic Acids Res. 21:4330–4338. 12. Evans, D. B., K. Brown, M. R. Deibel, Jr., W. G. Tarpley, and S. K. Sharma.
1991. A recombinant ribonuclease H domain of HIV-1 reverse transcriptase that is enzymatically active. J. Biol. Chem. 266:20583–20585.
13. Finston, W. I., and J. J. Champoux. 1984. RNA-primed initiation of Molo-ney murine leukemia virus plus strands by reverse transcriptase in vitro. J. Virol. 51:26–33.
14. Fu, T.-B., and J. Taylor. 1992. When retroviral reverse transcriptases reach the end of their RNA templates. J. Virol. 66:4271–4278.
15. Fuentes, G. M., L. Rodrı´guez-Rodrı´guez, P. J. Fay, and R. A. Bambara. 1995.
on November 9, 2019 by guest
http://jvi.asm.org/
Use of an oligoribonucleotide containing the polypurine tract sequence as a primer by HIV reverse transcriptase. J. Biol. Chem. 270:28169–28176. 16. Furfine, E. S., and J. E. Reardon. 1991. Human immunodeficiency virus
reverse transcriptase ribonuclease H: specificity of tRNALys3
-primer exci-sion. Biochemistry 30:7041–7046.
17. Furfine, E. S., and J. E. Reardon. 1991. Reverse transcriptase-RNase H from the human immunodeficiency virus. Relationship of the DNA polymerase and RNA hydrolysis activities. J. Biol. Chem. 266:406–412.
18. Georgiadis, M. M., S. M. Jessen, C. M. Ogata, A. Telesnitsky, S. P. Goff, and W. A. Hendrickson.1995. Mechanistic implications from the structure of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase. Structure 3:879–892.
19. Gilboa, E., S. W. Mitra, S. Goff, and D. Baltimore. 1979. A detailed model of reverse transcription and tests of crucial aspects. Cell 49:347–356. 20. Gopalakrishnan, V., J. A. Peliska, and S. J. Benkovic. 1992. Human
immu-nodeficiency virus type 1 reverse transcriptase: spatial and temporal rela-tionship between the polymerase and RNase H activities. Proc. Natl. Acad. Sci. USA 89:10763–10767.
21. Hizi, A., S. H. Hughes, and M. Shaharabany. 1990. Mutational analysis of the ribonuclease H activity of human immunodeficiency virus reverse tran-scriptase. Virology 175:575–580.
22. Hostomsky, Z., Z. Hostomska, G. O. Hudson, E. W. Moomaw, and B. R. Nodes.1991. Reconstitution in vitro of RNase H activity by using purified N-terminal and C-terminal domains of human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 88:1148–1152. 23. Hottiger, M., V. N. Podust, R. L. Thimmig, C. McHenry, and U. Hu¨bscher.
1994. Strand-displacement activity of the human immunodeficiency virus type 1 reverse transcriptase heterodimer and its individual subunits. J. Biol. Chem. 269:986–991.
24. Huber, H. E., J. M. McCoy, J. S. Seehra, and C. C. Richardson. 1989. Human immunodeficiency virus 1 reverse transcriptase: template binding, processiv-ity, strand displacement synthesis, and template switching. J. Biol. Chem. 264:4669–4678.
25. Huber, H. E., and C. C. Richardson. 1990. Processing of the primer for plus-strand DNA synthesis by human immunodeficiency virus 1 reverse tran-scriptase. J. Biol. Chem. 265:10565–10573.
26. Kotewicz, M. I., C. M. Sampson, J. M. D’Alessio, and G. F. Gerard. 1988. Isolation of cloned Moloney murine leukemia virus reverse transcriptase lacking ribonuclease H activity. Nucleic Acids Res. 16:265–277.
27. Luo, G., L. Sharmeen, and J. Taylor. 1990. Specificities involved in the initiation of retroviral plus-strand DNA. J. Virol. 64:592–597.
28. Omer, C. A., and A. J. Faras. 1982. Evidence for involvement of an RNA primer in initiation of strong-stop plus DNA synthesis during reverse tran-scription in vitro. Cell 30:797–805.
29. Oyama, F., R. Kikuchi, R. J. Crouch, and T. Uchida. 1989. Intrinsic prop-erties of reverse transcriptase in reverse transcription. Associated RNase H is essentially regarded as an endonuclease. J. Biol. Chem. 264:18808–18817. 30. Prasad, V. R. 1993. Genetic analysis of retroviral reverse transcriptase struc-ture and function, p. 135–162. In A. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. 31. Prasad, V. R., and S. P. Goff. 1989. Linker insertion mutagenesis of the
human immunodeficiency virus reverse transcriptase expressed in bacteria: definition of the minimal polymerase domain. Proc. Natl. Acad. Sci. USA 86:3104–3108.
32. Pullen, K. A., and J. J. Champoux. 1990. Plus-strand origin for human immunodeficiency virus type 1: implications for integration. J. Virol. 64: 6274–6277.
33. Pullen, K. A., L. K. Ishimoto, and J. J. Champoux. 1992. Incomplete removal of the RNA primer for minus-strand DNA synthesis by human immunode-ficiency virus type 1 reverse transcriptase. J. Virol. 66:367–373.
34. Randolph, C. R., and J. J. Champoux. 1994. The use of DNA and RNA oligonucleotides in hybrid structures with longer polynucleotide chains to
probe the structural requirements for Moloney murine leukemia virus plus strand priming. J. Biol. Chem. 269:19207–19215.
35. Rattray, A. J., and J. J. Champoux. 1987. The role of Moloney murine leukemia virus RNase H activity in the formation of plus-strand primers. J. Virol. 61:2843–2851.
36. Rattray, A. J., and J. J. Champoux. 1989. Plus-strand priming by Moloney murine leukemia virus: the sequence features important for cleavage by RNase H. J. Mol. Biol. 208:445–456.
37. Schatz, O., J. Mous, and S. F. J. LeGrice. 1990. HIV-1 RT-associated ribonuclease H displays both endonuclease and 39-59exonuclease activity. EMBO J. 9:1171–1176.
38. Schultz, S. J., S. H. Whiting, and J. J. Champoux. 1995. Cleavage specific-ities of Moloney murine leukemia virus RNase H implicated in the second strand transfer during reverse transcription. J. Biol. Chem. 41:24135–24145. 39. Shinnick, T. M., R. A. Lerner, and J. G. Sutcliffe. 1981. Nucleotide sequence
of Moloney murine leukemia virus. Nature (London) 293:543–548. 40. Shoemaker, C., S. Goff, E. Gilboa, M. Paskind, S. W. Mitra, and D.
Balti-more.1980. Structure of a cloned circular Moloney murine leukemia virus DNA molecule containing an inverted segment: implications for retrovirus integration. Proc. Natl. Acad. Sci. USA 77:3932–3936.
41. Smith, D. B., and K. S. Johnson. 1988. Single step purification of polypep-tides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67:31–40.
42. Smith, J. K., A. Cywinski, and J. M. Taylor. 1984. Specificity of initiation of plus-strand DNA by Rous sarcoma virus. J. Virol. 52:314–319.
43. Smith, J. S., K. Gritsman, and M. J. Roth. 1994. Contributions of DNA polymerase subdomains to the RNase H activity of human immunodefi-ciency virus type 1 reverse transcriptase. J. Virol. 68:5721–5729.
44. Smith, J. S., and M. J. Roth. 1992. Specificity of human immunodeficiency virus-1 reverse transcriptase-associated ribonuclease H in removal of the minus-strand primer, tRNALys3. J. Biol. Chem. 267:15071–15079.
45. Smith, J. S., and M. J. Roth. 1993. Purification and characterization of an active human immunodeficiency virus type 1 RNase H domain. J. Virol. 67:4037–4049.
46. Steitz, T. A., S. J. Smerdon, J. Ja¨ger, and C. M. Joyce.1994. A unified polymerase mechanism for nonhomologous DNA and RNA polymerases. Science 266:2022–2025.
47. Tanese, N., and S. P. Goff. 1988. Domain structure of the Moloney murine leukemia virus reverse transcriptase: mutational analysis and separate ex-pression of the DNA polymerase and RNase H activities. Proc. Natl. Acad. Sci. USA 85:1777–1781.
48. Tanese, N., A. Telesnitsky, and S. P. Goff. 1991. Abortive reverse transcrip-tion by mutants of Moloney murine leukemia virus deficient in the reverse transcriptase-associated RNase H function. J. Virol. 65:4387–4397. 49. Telesnitsky, A., and S. P. Goff. 1993. RNase H domain mutations affect the
interaction between Moloney murine leukemia virus reverse transcriptase and its primer-template. Proc. Natl. Acad. Sci. USA 90:1276–1280. 50. Telesnitsky, A., and S. P. Goff. 1993. Two defective forms of reverse
tran-scriptase can complement to restore retroviral infectivity. EMBO J. 12:4433– 4438.
51. Varmus, H., and P. Brown. 1989. Retroviruses, p. 53–108. In D. E. Berg and M. M. Howe (ed.), Mobile DNA. American Society for Microbiology, Wash-ington, D.C.
52. Verma, I. M. 1977. The reverse transcriptase. Biochim. Biophys. Acta 473: 1–38.
53. Whiting, S. H., and J. J. Champoux. 1995. Strand displacement synthesis capability of Moloney murine leukemia virus reverse transcriptase. J. Virol. 68:4747–4758.
54. Yang, W., W. A. Hendrickson, R. J. Crouch, and Y. Satow. 1990. Structure of ribonuclease H phased at 2 Å resolution by MAD analysis of the selenome-thionyl protein. Science 249:1398–1405.