0022-538X/92/052973-09$02.00/0
Copyright ©1992, American Society for
Microbiology
Effect
of
Genomic Location
on
Expression
of
1-Galactosidase
mRNA
Controlled by the
Herpes
Simplex Virus
Type
1 UL38
Promoter
SHERYL A. GOODART, JOHNF. GUZOWSKI, MARCIA K. RICE,ANDEDWARD K. WAGNER*
DepartmentofMolecular BiologyandBiochemistry, Universityof California Irvine, Irvine, California 92717 Received 14 November 1991/Accepted5February 1992
To examine the effect of genomic location on the details ofexpression of selected herpes simplex virus
promoters,wehaveconstructedrecombinationvectorsfor placing suchpromoterscontrollingthe
1-galactosi-dasereportergeneintotworegions oftheviral genomelackinganynearbypromoterorregulatory elements.The first vector generates the promoter-B-galactosidase reporter gene inverted within the locus of the gC
(UL44) translational reading frame; the second replaces the LAT promoter and the first 600 bases of the
primary transcript in both copies of the RL region. These locations were chosen to obviate any possible
influenceofupstreambutnoncontiguous heterologousorhomologous DNAsequenceelementsuponpromoter activity.When thereportergenecontrolledby the strict late (-y) UL38promoterwasplaced inthegClocation, itwas significantlyless active than in its normal location; in contrast, promoteractivitywas comparable to
wild-typevalues when the promoterwasrecombined into the RL region. The low levelofactivity in the gC locationcould bepartially alleviated by theincorporation of additionalDNAsequencesupstreamof theUL38 promoter.Despitetheeffect of genomiclocationuponthelevelof expression, the kineticsof expressionineither
locationmirrors the wild-typeUL38 strict late kineticsof expression. Finally, weuseddeletional analysis to demonstrate thatno morethan29 basesofDNAsequence5' of the mRNAcapsitearerequiredforpromoter activity in either location; this result is consistent with earlier results of transient-expression assays and indicates that the UL38 promoter shares general features with other strict late (-y) herpes simplex virus promoters.
Transcripts encoded by herpes simplex virus type 1
(HSV-1)areexpressed duringthelytic replication cycle ina
sequential cascade pattern (14). Regulation of this expres-sionpatternoccursprimarilyatthe level oftranscription (21, 26, 29, 31). Transcripts encodingthe fiveimmediate-early(a)
genes are the first to be expressed; these gene products,
especiallyICP4 (ao4), are required for expression of subse-quentclasses of HSV-1genesduringlytic expression. Early
ordelayed-early
(pi)
genesarethesecondgroupofgenesto be expressed. Generallythe polypeptide products of thesegenesareinvolved inreplication of viralDNA; their
expres-siontendstoreachmaximal levels around thepeakof DNA
replication and then decrease (31). Late transcripts are maximally expressed only after the onset of viral DNA
replication. They can begrouped into leaky-late (,3yor
yl)
genes and true or strict late (y or _Y2) genes; the difference between these two subclasses is in the stringency of the
requirement for DNA replication. Transcripts encoding
P-y
genes are expressed at low but readily detectable levels
before DNA synthesis, whereas -y transcripts evidence a strict requirement for DNA replication for any significant
expression (10).
The promoterscontrollingthe -yor strict late genes have been the subject of some investigation, especially those
controllingthegH (UL22), UL38, gC(UL44),and
Us11
genes(11, 13, 16, 17, 22, 24). Although these promoters do not evidencearequirementforviral DNAreplicationfor
expres-sion in transient-expression assays, the extent of the
pro-motersdelineatedgenerallyagreeswiththosedeterminedby
generation of recombinant virusescontainingmodified
pro-* Correspondingauthor.
motersequences. Currentdataindicate that ypromotersare
quitelimited inlength, extendingfromarecognizableTATA
homology 28 to 30 bases 5' of the mRNA cap site and
includingsequences nearthetranscriptinitiationsite
extend-ing to 10to 20 bases 3' of thispoint.
The limited number ofDNAsequence elements
responsi-ble for the control ofytranscription is of interest becauseof
its potential utilityin saturation mutagenesis studies and as
probes for theidentification of transcription factors impor-tant in late gene expression. As noted, to date the most clearly recognizable promoter element found in these
pro-moters is the TATAhomology. Since this element is such a
notablefeatureofall-ypromotersstudied, it isreasonableto speculate that a protein factor(s) which recognizes this element might play a primary role in the expression of y
genes. Further, since-ygenes requireviral DNAreplication
forexpression,it is plausibleto speculatethatsuchafactor
might be altered in infectiontoenable it tobetterrecognize promoters.
Despitetheubiquityof-y promoterTATAboxes,there is some controversy concerning the actual sequence
require-ments for function. Johnson and Everett reported that in
transient-expression assaystheycould change theearlygD
(U,6)
promoterintoan operationallystrict latepromoteron aplasmid containing the HSVori,
by truncating it at -33, leaving the TATA element as the only recognizablepro-moterelement(16).Morerecently, SteffyandWeirreported
thatthree TATA elements derived from theao4, Lthymidine
kinase (TK; UL23), and y gC (UL44) promoters could substitute for the y gH(UL22) TATA elementina
recombi-nantvirus, although the levels ofexpression differed signif-icantly dependingupon the exactelement used (24). These results argue against a strict sequence requirement for a 2973
on November 9, 2019 by guest
http://jvi.asm.org/
functional -y TATA box. However, Homa et al. concluded that the TK (UL23) promoter-leader (-37 through +57)
possessing
the intact TATA box(ATATTAA)
could notsubstitute for the gC promoter-leader
(-146 through
+124) inrecombinant virus(11,
12). NogCmRNA wasexpressed from theTKpromoter-leader-gC
fusion until the TK TATA element was mutated toGTATAAA,
which more closely resembles the gC sequence, TATAAATT. This mutated TATA box generated 10% ofwild-type (wt)
levels of gC mRNA.Although
results of studies with recombinant virusesbearing
mutated -y promoterregions generally
agree with results oftransient-expression
assays, theseexperiments
have beencarriedout atthe normallocation of the promoter inquestion (11,
12,24).
This allows thepossibility
thatnoncontiguous
DNA sequence elements further upstreamof the TATA box will influence mRNAexpression.
Further, suchexperiments
require
that the promoter inquestion
control expressionofa genewhich is nonessential for viralreplication
in cell culture. We havedeveloped
two recombi-nation vectors which allow the insertion of any HSV pro-motercontrolling
the3-galactosidase
indicator gene intotworegions
ofthe HSV-1 genome,onewithin the location of the nonessential gC(UL44)
gene and the other replacing aportion
ofthelatency-associated transcription
unitand pro-moterinthelong
repeat(RL)
region.Thesetwolocationsof recombination will obviate the influence of any specific promoter elementsoccurring
upstream of the "core" pro-moter and allow a rough estimate of the effect ofgenomicposition
onexpression.
In this system, thewttranscription
unit and control sequences of the gene whose promoter isbeing
studied are not altered and theexpression
of the wt mRNAprovides
an absolute internal standard for measure-ment.We have chosen the promoter
controlling
theUL38
gene as a useful model for the furtheranalysis
of -y promoters. This promoter iswithin a280-bp region
of DNA which also contains the promoter for the1B
UL37
mRNAtranscribed in theopposite
direction(2, 31).
These two promoters arecompletely separable
intransient-expression
assays. Se-quencesextending
from -29 to +10 relativeto the cap siteprovide essentially
fullexpression
uponviralsuperinfection,
and DNAextending
from -48to +99 basesissufficientfor theexpression
of a marker gene(13-galactosidase)
in a recombinant virus. Three T+A-richregions
occur in this latterregion:
AAATat -38, TTTAAAat-29,andTATAat-17.
Further, antibody-mediated
DNA supershift assays indicate that thecx4protein
interacts with DNAnearthe capsite; and,
finally,
the target for this interaction has sequencehomology
with promoter elements of theyy42
(UL49)
gene(6).
Inthecurrentreport,wedescribe the details of
expression
of UL38-controlled
1-galactosidase
in these recombinant virus systems. With the UL38 promoter, we found thatgenomic position strongly
influences the level butnot kinet-ics ofexpression.
Deletionalanalysisshows that in thevirus, the sequenceT'`TTAAA
between -29 and -23 of theUL38 mRNAcapsite is absolutely required fortranscription, but levelsofexpressioncanbereadilyincreasedbytheadditionoffurthersequence elements farther upstream.Thus,we can
clearly
differentiate sequence elements required for the kinetics ofexpression
from thoseimportant
ininfluencing
levels ofexpression.
The unusual nature of theUL38TATAhomology
supports the major body of experimental dataindicating
that aspecific
sequence ofthe -y TATAbox is of minorimportance
inmediating
transcription.MATERIALS AND METHODS
Cells and virus. HSV-1
(17syn')
was the parental strain usedto generate the recombinant viruses described below. Parental and recombinant virus infection of rabbit skin fibroblastsat amultiplicity of infection of 5 PFU per cellwasused to generate the RNA used for Northern (RNA) blots and RNase protection assays. Vero cells were used for plaque assays, and both cell types were used during
recom-binant virus screening and isolation. The cells were main-tained at 37°C under 5% carbon dioxide in Eagle minimum essential medium containing 5% cadet calf serum, 100 U of penicillin perml, and 100 ,ug of streptomycin per ml (3, 5,7, 30).
Enzymes. Restriction enzymes were purchased from Boehringer Mannheim Biochemical Corp.; Bethesda Re-searchLaboratories,Inc.;orNewEngland Biolabs. Restric-tionbuffers recommended by the suppliers were used for all digestions. T7 RNA polymerase and RNasin used in RNase protection were purchased from Promega Biotec, Madison, Wis. T4polynucleotide kinase (Bethesda Research Labora-tories) was used to 5' end label appropriate markers for RNase protection. Finally, Klenow enzyme used for both labeling and blunting the ends of DNA fragments was purchased from Boehringer Mannheim.
Recombinant DNA. pBR322 and pGEM plasmid deriva-tives were grown in Escherichia coli DH5a. Growth and purification of plasmids were essentially as previously de-scribed (1, 7, 18). Oligonucleotide linkers were purchased from Collaborative Research, Inc., Waltham, Mass. DNA fragments for probe generation were isolated by digestion with the appropriate restriction enzymes followed by elec-trophoresis and either electroelution from agarose or over-nightelution from acrylamideat37°C inasolution of 500 mM ammonium acetate, 10 mM magnesium acetate, 1 mM EDTA, 0.1% (wt/vol) sodiumdodecyl sulfate, and 10 pgof E. coli tRNA(Sigma) per ml. Probeswereuniformly labeled by random priming andsynthesis by using Klenow enzyme with [ct-32P]dCTP (3 Ci/,umol; Amersham).
RNAisolationand fractionation. Methodsfor isolation of RNAbyusing guanidinium isothiocyanate-hot phenol have been previously described (27, 28). RNA generated from infected cells was isolated at specific times postinfection (p.i.) and either useddirectlyastotal RNAorfirst fraction-ated into poly(A)+ and poly(A)- fractions by use of oli-go(dT)-cellulose (Collaborative Research) chromatography (27).
Phosphonoacetic acid (PAA) or thymine
1-0-D-arabi-nofuranoside (ara-T)wasusedtoinhibit viral DNA replica-tion. Cellswereincubated with PAA(300,ug/ml)
for30 min before infection or with ara-T (50 ,ug/ml) for 1 h before infection.Drugswerepresentduringviraladsorptionand for thetimes noted in thetext.Actinomycin D(10 ,ug/ml)wasusedto study thestability of the mRNAs of interest by its addition 6 h p.i. The infectionwasallowedtoproceedforanother 1or2h before RNAisolation.
Recombinant virus. All sequence numbers are based on thecompletesequenceof the17syn+ strain of HSV-1(19).A gCrecombinantvectorextendingfrom theSall site atbase 94853in thegC regionto theHindIIIsiteatbase98823was
constructed by using appropriate clones from the 17syn+ strain of HSV-1. This construct contains an intactgC
pro-moter. The 3.9-kbpXbaI-BamHI cassette of pCAL5Div17 (6)contains 48basesof theUL38 promoter 5' of the cap site and 99 bases ofUL38 leader fused tothe bacterial
on November 9, 2019 by guest
http://jvi.asm.org/
tosidase gene with a 4-base linker. It is terminated with the bidirectional simianvirus 40(SV40) termination-polyadeny-lation sequences. The BamHI site was blunted with Klenow enzyme, andthecassettewasligated into the
XbaI-EcoRV-digested gC
recombinant vector, replacing 22 bp of gC sequences. This construct places the chimeric gene in theopposite
orientation to and downstream of the gC cap and promoter. As constructed, both a truncated gC and a chi-meric3-galactosidase
transcriptcanterminate at the bifunc-tional SV40 termination-polyadenylation signal. This con-structcontainsa360-bpXbaI-Kp4nI
cassettecontaining UL38promoter-leader
sequencesand the 5' portion of the P-galac-tosidase gene. Promoterconstructswith less DNA 5' of the cap site were constructed by replacement of this cassette withappropriate
XbaI-Kpnl
cassettes from the UL38 pro-moter constructs described elsewhere (6). A larger UL38promoter-,3-galactosidase
recombinant vector including all theUL37
and UL38 promoterregionsdriving0-galactosidase
wasgenerated
by first inserting an XbaI oligonucleotide linker at the SalI site near the UL37-CAT junction inpCALSDiv
and thencloningthisXbaI-KpnI
cassette into the recombinantvector.The
generation
of the recombinant virus FLA5, which contains 48 bases of the UL38 promoter and the 99-base leader fusedtothe,B-galactosidasegene recombined into theRL
regions
of the genome substituting for the latency-associatedtranscript
(LAT) promoter and 5' sequence, has been describedpreviously (6). Itwasconstructed by using a recombination vector plasmid in which 1,800 bp of HSV DNA of the RLregion
containing the LAT promoter and 5' sequencesof theprimary
latency-associatedtranscript werereplaced
with the same XbaI-BamHI fragment containing the indicator gene as used in the gC recombination vectorplasmid
described above. OtherRLrecombinant virus con-structs weregenerated
by replacing either theXbaI-KpnI
cassette or the XbaI-BamHI cassette of the recombinant vectorwithappropriate
modifications or in reversed orien-tation in theRL
region.
Recombinant virus was generated by first releasing the HSV-1 sequences in the plasmidvectors bydigestion with
appropriate
restriction enzymes and then cotransfecting cloned DNA sequences with infectious17syn'
DNA into rabbit skinfibroblasts. Single-virus plaquesderived bylim-iting
dilutionwere screened in 96-well plates by hybridiza-tion of transfechybridiza-tionaliquots
with32P-labeled
0-galactosidase
probe.
Wells containing recombinant viral DNAwere thenplaque purified
atleastthreemoretimesby repetition of thefollowing procedure.
Isolatedplaqueswerepickedfrom agaroverlays
oflimiting
dilutions of virus infecting Vero cells into 96-well plates containing rabbit skin fibroblasts and screenedby hybridization.
Theresulting isolates were thenanalyzed by
Southern blot. Purified isolates containing thecomplete promoter-p-galactosidase
construct inserted into thegC region
forgC
recombinant virusorinto both the IRL andTRLregions
for the RL recombinant virus were used in this report.Plaque-purified
isolates from different transfec-tionswere takenfor each construct, and thelevel of expres-sion of the chimeric3-galactosidase
genewas the same asassayed by
colorimetric methods. Theequivalency of levels of RNAexpression
from different recombinants bearingdiffering lengths
ofupstreampromoter sequenceorindiffer-ent orientations confirmed that the expression seen was a result of the
specific
recombinations characterized andwas not due to some nonspecific mutation in another region ofthe viral genome.
RNase protection assays. RNase protection assays have
been described elsewhere (4, 9). pGEM templates were used with T7 polymerase (Promega Biotec) by following instruc-tions supplied to generate the RNA probes. The probe was
labeled with 100
,uCi
of[32PJUTP
(800 Ci/mmol; Amersham) per,ug
of template DNA. The UL38 protection probe was aHhaI fragment generated from the
UL38-,B-galactosidase
chimeric recombinant virus construct. The HhaI fragment spansthe region of DNA from 5 bp 5' of the UL38 cap to 28 bpwithin the
,-galactosidase
leader. This probe protects 99 bases of wild-type UL38 and 127 bases of UL38-,B-galactosi-dase recombinant mRNA. A control dUTPase protection probe was aBglII-Kjpnl
fragment extending 62 bp 5' of the capto 95 bp3' of the cap. This dUTPase probe protects 95 bases of 17syn'wild-type dUTPase mRNA.More than 95% of total RNase protection probes synthe-sized with T7 polymerase were full length on the basis of results with analytical gels. A total of 1.5 x
106 cpm
(Cerenkov) of probe was hybridized with 10 to 20 pLg of appropriate RNA samples in 50,ulof80% formamide-40mMpiperazine-N,N'-bis(2-ethanesulfonic acid) (PIPES; pH
6.4)-400mM
NaCl-lmM EDTA at60°C
for 11 h. Themix-turewasthendigested by addition of 400,ulof RNase T1 (2 ,ug/ml; Sigma) in 10 mM Tris (pH 7.5)-300 mM
NaCl-SmM
EDTAfor 1 h at30°C.
The digestion mix was next treated with 60 pLg of proteinase K (Sigma) at37°C
for 15 min and phenol-chloroform extracted. RNase-resistant material was fractionated on an 8% acrylamide-8 M urea sequencing gel with DNA size standards.Northern blot analysis. RNA transfer blots were hybrid-ized and washed essentially as described previously (6, 31). An 8-ml volume containing 4 x
107
cpm (Cerenkov) of radiolabeled random-primed DNA in the presence of 50% formamide, 0.4 M Na+, 0.1 M HEPES (N-2-hydroxyeth-ylpiperazine-N'-2-ethanesulfonic acid; pH 8.0), 0.005 M EDTA, and Denhardt's solution containing 100,ug
of dena-tured calf thymus DNA per ml was used at50°C
for 40h.RESULTS
Placement ofUL38promoter-controlled expression marker in different locations in the HSV-1 genome. To eliminate the influence of transcription signals upstream of the UL38 core promoter, we generated recombinant viruses in which the
promoter-indicator gene cassette was placed in the inverted
orientation in the gC locus. The general structure of such
recombinants is shown in Fig. 1A; the 5' terminus of the UL38 promoter is bounded by anXbaI site at base 97669. This is 250 bases 5' of the cap site of the 730-base -y mRNA encoding the UL45 gene, which is coterminal with the 3' end of gC (8). The genomic structure and purity ofrecombinants were confirmed by Southern blots of viral DNA. As shown in Fig. 1A, the 3,600-bp Sall fragment was replaced by the 7,440-bp fragment containing the 3-galactosidase gene, var-ious portions of the UL38 promoter, and the SV40 termina-tion-polyadenylation sequences described in Materials and Methods.
The bidirectional activity of the SV40 polyadenylation signal was confirmed by analysis of RNA expressed by recombinant viruses containing the chimeric UL38-I-galac-tosidase reporter gene in the gC region. Figure 2A showsan
example in which Northern blots of poly(A)+ RNA from
cells infected with the UL38 promoter construct from -48 to +99 expressed a truncated 1.5-kb portion of gC mRNA (track ii) compared with the 2.7-kb wt gC mRNAexpressed in cellsinfected with the 17syn' strain (wt, track iii).
on November 9, 2019 by guest
http://jvi.asm.org/
SoIl-3600bp
Xba IRL IRSUS TRS
I J
UL38,SoSoUTPose
SV40(A+)
So 1500bpj, 3400bp Xba Sa
(948531) (9B1 (97669)(98422)
Sall* -7440bp
aQ)J3? bz4P" aR
VD C
lX lv eDC
-23
-9.4
-6.6 j-44
-23 !-2.0
23
-9.4
-66
-4.4
B. TRL
IIR IRS -Rs
Xbo Xba Xbo Xbc
-_1 I;
poUUSalSal-- UL38 "-l So! Us
C-8767bp f E-6385bp
a20 <O~ 017TRL
X
'a-Sal 3400bp -Ps-, Xba Sa' C)
(5469) Xba (10636) 114232) / (7707)
SoIC -10,851 bp / IR, l9.
Sal -Pst- 3400bp So!
(114517) Xba (120,902)
(118659)
SolE*-8468bp
so
,-ga:probe
-4C
2.3-2.0
-gC probe
LATprobe 7_
934 -6; 6
-44
-2 3
-20.
rXC,/;51 ::! CXD/ O'
FIG. 1. HSV-1 recombinant viruses. The location and precise sequence numbers where the reporter gene-promoter constructs were
inserted into the HSVgenome areindicatedin themaps.(A) Schematic representationof theexpectedstructureof recombinantscontaining the UL38 promoter-controlled 1-galactosidase gene recombined into the gC region of the HSV-1 genome. Insertion of the promoter-n-galactosidase constructsgenerates aSalI fragment (7,440 bp) which is largerthan thewtfragment (3,600 bp). Below themapareshown diagnostic Southern blotsdemonstrating the insertion ofthe ,-galactosidase fragment. DigestionwithSall generates the expected-sized fragmentfor therecombinant viruseswhich hybridizestoboth 1-galactosidase-specific andHSV-1-specific probes.The latterprobewas a
945-bp PstI-SalI fragment (correspondingtobases 97477through98422of thecomplete17syn'sequence[191)cloned from thegC regionof
wtHSV-1 DNA. Bycontrast, wtviral DNA digested withSallgeneratestheexpected fragment which hybridized onlytothegC-specific probe. (B) Schematic representation of theexpected structureoftheRLrecombinants with theUL38 promoter-controlled 3-galactosidase
generecombinedinto boththeTRLandtheIRL regions ofthe HSV-1genome.Insertionintobothregionsgeneratestwonewfragmentsupon digestionwithSall:SalC*(10,851bp)andSalE* (8,468 bp).ThesefragmentsarelargerthanthosegeneratedbySalldigestionofwtHSV-1 DNA:fragments SaiC(8,768 bp)andSalE(6,385 bp), respectively.Therecombinantfragments hybridizetoboth 3-galactosidase-specificand
wtHSV-1-specific probes.The latterprobewas a605-bpHpaI-SalI fragment (correspondingtobases 120297through 120902) from theRL regionofwtDNA.Digestionwith XbaI andSalI generateda3.9-kbfragmentfrom theRLrecombinants.Thisfragment hybridizeswith both
3-galactosidaseand HSV-1probes.ThesamedigestofwtDNAgenerated6.3- and 5.2-kbfragments,whichweredetectableonlywith the HSV-1-specific probe.
Hybridization of blots with probes specific for UL38
mRNA revealed both the normal 1.9-kbUL38mRNA and a 3.4-kb chimeric mRNA containing the 99 bases of UL38
leader and ,B-galactosidase sequences, whereas RNA from
wt-infected cells expressed only UL38 mRNA (Fig. 2B,
tracks i andii). Hybridization of blots with ,B-galactosidase-specific probes reveals only the 3.4-kb mRNA (data not shown).
TheUL38
promoter-p-galactosidase
reporter cassettewas also recombined in both orientations into the RL regionof the HSV-1 genome by the substitution of the proximal promoterand5'regionsof thelatency-associatedtranscrip-tion unit.Examples of Southernblots of such recombinants
showingrecombination into both repeatsareshown inFig. 1B; recombination results in thegeneration of the 10.8-kbp SalI fragment C* and the 8.5-kbp Sall fragment E* deriva-tivescontainingthe
P-galactosidase
reportergeneconstruct replacingthe8.8-and 6.4-kbp parental fragments.Recombination of the reporter construct into the RL
region also yielded recombinant virus which exhibited
sig-nificant expression of the chimeric
UL38-p-galactosidase
mRNApromoteras wellas normal UL38mRNA (Fig. 2B, track iii). These recombinants expressed normal-sized gC
mRNA (Fig. 2A, tracki). Inotherexperiments (results not
shown)wedetermined thatrecombination ineither regionof theHSVgenome had noeffectuponthesizeor theamount ofaOmRNA,whichterminatesjust3' of the site ofinsertion
ofthereporter constructinto the RL region.
Amounts ofreportergenemRNAexpressedareinfluenced
by the genomic position of the promoter controlling them.
AlthoughUL38 promoter-controlled ,B-galactosidase mRNA wasefficiently expressed fromrecombinantviruses bearing
thecassetteeither in thegClocationorin theRL regions,the levels of chimeric
UL38-I-galactosidase
mRNA seen in Northern blotswasalwaysgreaterfor the latterconstructs. We confirmed and quantitated this difference in mRNAlevels by use of RNase protection assays with an RNase
probe extending from a HhaI site in the p-galactosidase
sequencesof the reportercassette28 bases 3'ofthejunction
of theUL38leaderto aHhaIsite 5' of the mRNAcap site (see Materials and Methods). Chimeric
UL38-,B-galactosi-dase mRNAprotected 127 bases of thisprobe,whereaswt UL38 mRNA protected 99 bases. Since the probe is
uni-formly labeled and since protected fragments have
essen-tianly
thesamesize for bothmRNAs,adirectcomparisonof the intensity of RNase-resistant hybrids results in a directmeasureof the relativeamountsof both mRNAs in infected
cells in which the amount ofwt UL38 mRNA provides an internal standard. Further, since the structure of the chi-meric mRNAwasthesamefor allrecombinants, any
differ-encesin levels of mRNAmustreflect differences in levels of
expression and not differential mRNA stability or other factors.
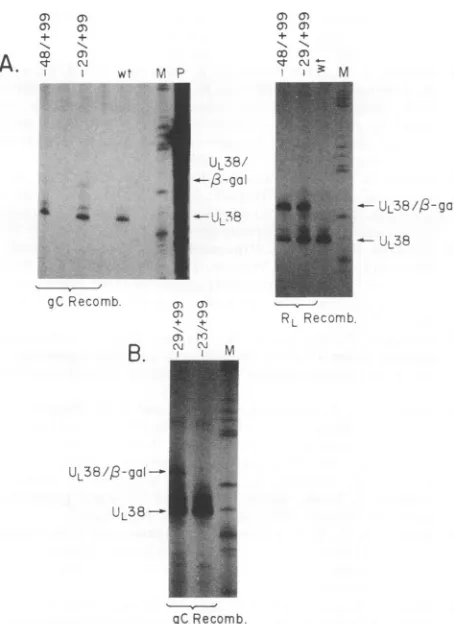
Our datawereidentical forUL38promoterscontaining48 and 29 bases of DNA sequence upstream of the cap site. Results ofatypical experimentareshown inFig.3;the ratio
A.
TRL UL
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.97.535.76.285.2]o J
U L
3
03R
5Kb-.-(I) (ii) 1
030
034-A.
cXct CMj
P MIM2
03
I II
P M2
aw~~~~~~1
:,WI" 0
2Kb
UL38/'.2g-r3l-~
UL'38
-U[L38probe (i) ( ii (lii)
gCprobe
FIG. 2. Northern blot analysis of chimeric and gC mRNAs expressed during infection with recombinant viruses. Samples of poly(A)+ RNAwereisolated from rabbit skin fibroblasts 4 h after infection (hpi) with recombinant orwtvirus,fractionated, blotted, and hybridized. (A) Representative Northern blot probed with an
896-bp EcoRI-EcoRV DNA fragment specific for gC (UL44) mRNA (correspondingto bases96524 through 95628). The 1.5-kb mRNA expressed by the recombinants (trackii) indicates that recombina-tionintothe gC region results in truncation of the gCmRNA. Both the RL recombinant (track i) and parental wt (track iii) viruses
express full-length gC mRNA (2.7 kb), as expected. (B) Repre-sentative Northernblot of gC and RL recombinantsalong with their parental wt RNAprobedwith theXbaI-KpnI fragment from the -48/+99-,-galactosidaseconstruct.Thisfragment hybridizesto wt
UL38 mRNAand to ,-galactosidase-containing mRNA. All three virustypesexpresswtUL38 mRNA(1.9 kb), whereas only thetwo
recombinantsexpresstheUL38-3-galactosidasemRNA (3.4 kb).
of chimericto wtmRNAwas determineddensitometrically in two to four separate gels. Ratios varied ±10% between gels.Chimeric
U_38-3-galactosidase
mRNAexpressed from the UL38 promoter in the gC region recombinants wasreproduciblypresent at20%of thelevel ofwtUL38mRNA
(tracks i and iii). Incontrast, recombinant viruses with the samepromoter constructrecombined intoeitherorientation in the RL region expressed essentially twice as much chi-meric mRNAaswtUL38mRNA(tracksvandvi).Since the recombinantpromoter-reporter geneconstructispresent in
twocopies in the RLconstructs, ourdata indicate that the UL38 promoter is as active in the RL region as it is in its normal location.
Thelower level ofexpressionof theUL38promoterinthe
gClocation could bepartiallyalleviatedbytheincorporation
of DNAsequencesextending beyond48 bases of the mRNA cap site. Thus, a construct containing 232 bases of DNA
sequence inserted upstream ofthe 48-base UL38promoter resulted ina two- tothreefold increase in chimeric mRNA levels (Fig. 3, tracks ii and iv). This additional sequence
contained the DNAsequencesextendingfrom thecapsite of theUL37 mRNA, throughits promoter, andincludingUL38
sequences5' of -48. Onceinserted, itreconstructed thewt
UL37-UL38 juxtaposed promoters with the UL38promoter
linked to ,3-galactosidase.
Weconfirmed thevalidityofourmeasurementsof mRNA levels as an index of promoter activity by comparing the
stabilityof chimeric
UL38-3-galactosidase
mRNAwith that ofthewttranscriptby using actinomycinchaseexperiments.Theratios ofwt tochimeric mRNA donotchange following
19*.-UL4 UL38/A -gal
i)38
(1)(11) OIO1 Ov) (v)(VI)
gC RL
Recombinant
B-48/+~48/99RL
B.~ ~~~~~~~~~~
ml _:i
tX I- UL38/i3-gal
6 6+1h hpi hpi Actino
FIG. 3. Quantitation of the levelofchimeric
UL38-I-galactosi-dase RNAexpressionin recombinantvirus. Total RNAwasisolated
from rabbit skin fibroblasts and hybridized to theHhaI
UL38-1-galactosidase probedescribed in Materials and Methods. Thisprobe
protects 99bases ofwt UL38 mRNA and 127 basesof chimeric UL38-P-galactosidasemRNA. Afterhybridizationand RNase diges-tion, the RNase-resistant material was fractionated on an 8%
denaturing acrylamide gel with EcoRI-Hinfl- (Ml) and HaeIII (M2)-digested pBR322 DNA fragments as size markers. (A) Ten micrograms (tracks i, ii,v,andvi)or20p.g(tracksiii andiv)of RNA isolated 6 h p.i.was used for eachvirus. Shown areRNAprobe fragments protected by RNAisolated fromcells infectedwithtwo
gCrecombinant viruses(-48/+99,tracks i andiii;-280/+99,tracks
ii and iv) and two RL recombinant viruses [-48/+99, track vi; -48/+99(R)in which the whole promoter-reportergeneconstructis reversed, trackvJ. A lane containing 600,000 cpm (Cerenkov) of undigested probe(P)wasincludedtocheck thelabeling efficiencyof theprobe.The size of theundigested probeis 197bp owingtothe
presence of the polylinkersequence. (B) RNAwasisolated from
cellsinfected with the-48/+99 RLrecombinant virusat6 hp.i.and
again after a 1-h treatmentwith 10 p.g ofactinomycin D per ml starting6 hp.i.The markers Ml and M2werethesame asdescribed above.
60to120minofchaseafter 6hof infection with recombinant virus; an example of a 60-min chase is shown in Fig. 3B. Therefore, the incorporation of bacterial 3-galactosidase
sequences in the reporter mRNA does not lead to any
significantincreaseordecrease in thestabilityof chimericas
comparedwithwtUL38mRNAwhich would interfere with
Ij(9)
QO cy)
A 4- 4-YCl}
A.
ODC
52Kb-..*
-4,I
4
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.76.282.80.250.2] [image:5.612.318.554.83.437.2]t-) U a
A -48/t99 -48/+99 B + + +i
U~8f-AI.
B.~
*
R of chmeoCcDac m
M M M 3'm 13
expressionToa.RAa m in
UL38/a-9a1-
E
t |t~~~~~~~~~~-
I-
JPose
denangacla gl w eIds P D
UL38-__Ii
2 6 12 2 6 12
[image:6.612.327.552.74.334.2]hp.
FIG. 4. Timecourse of chimeric UL38-B-galactosidase mRNA expression.Total RNAwasisolatedatvarious timesafterinfection, hybridized toanRNaseprotection probe,and subjectedtoRNase treatment, and the protected specieswerefractionated on an 8% denaturing acrylamide gelwithHaelll-digestedPBR322 DNA frag-ments as size standards (lanesM). (A) Total RNAs isolated from cells infected with either the-48/+99gCortheRLvirusat2, 6,and 12 hp.i. werehybridized tothe HhaIUL38-4-galactosidase probe described inMaterialsandMethods andin thelegendtoFig.3.(B) Total RNAs isolated fromcells infected withwtvirusorwith the -48/+99 gCorRL virusesat4and 6hp.i.werehybridized with the v dUTPase (IJL5O)-specific RNase protection probe described in Materials and Methods, which protects 95 bases ofwt dUTPase mRNA.
the use of the ratio ofchimeric to wt as a measure of the actual amounts of these mRNAs expressed in the infected cell.
Genomic location has no effect upon the kinetics ofUL38 promoter activity. We next examined the influence of ge-nomic position on the kinetics of UL38 promoter activity. TheexpressionofwtUL38 mRNA conformstostrict late
(y)
kinetics; thusexpressionof this mRNA absolutely requires viral DNAreplication (2, 31). Despite the variability in the levels of chimeric mRNA expressed from this promoter in the different recombinants, we found that its expression kinetics wereessentially thesamewhenit wasrecombined into either thegCorRLlocation in the genome. Results ofatypical experiment are shown in Fig. 4. RNA isolated at
various times after infection withwtorrecombinant viruses
wasanalyzed for the presence of UL38 and chimeric UL38-,-galactosidase mRNA. NoUL38or chimeric mRNA from recombinants in gCorin theRLregion could be detectedat
2 hp.i.Incontrast,these mRNAswerereadily detectable by 6 to 12 h p.i. when DNA replication is occurring at high rates. As acontrol, RNaseprotection assays with aprobe specific for the
P
class dUTPase (UL50) mRNA yielded relatively high levels at 2to 4 hp.i., with significantly less RNArecoverable from cells by 6to8hp.i.Sample data for4and8 hp.i. RNA are shown in Fig. 4B.
Astrictrequirement for DNA replication was also shown by using DNA synthesis inhibitors. In the presence of either 50 ,ug ofara-T perml or300 ,ugof PAA per ml, no wt or chimeric mRNA was expressed (Fig. SA). By contrast, similar samples of RNA hybridized with the
Pi
dUTPasemRNAprobeyielded levels as highas orhigher than those in
the untreated controls(Fig. 5B).
BasalUL38 promoteractivity requires sequences extending
nofarther than 29 bases5' of the transcript cap site. We next
A. -48 2+99 -280/+99
cI--<0
U 38/
U,38
gC Recomt
B.
dT:Pose
--48/+99
r-1-~~
M l
| :
-s:.E r
I
< 0-u CLq<
40 q JUL38/0901
L_ Recomb
-48 /+99 -280/+99
1
I..~~~~~~~~~~.
2-.r 2.0:- CM
hA r n) r C, 1 Cr
gCRecomb
FIG. 5. Effect of DNAreplicationontheexpressionof chimeric
UL38-p-galactosidase
mRNA.Total RNAwasisolated 6 hp.i. from rabbit skinfibroblasts infected with recombinant virusin thepres-ence orabsence(control[lanesC]) ofeither50 ,ugofara-T permlor
300 ,ug of PAA per ml. The RNAwas hybridized to an RNase protection probe, treated with RNase, and fractionatedon an 8% denaturing acrylamide gelwithHaeIII-digestedpBR322DNA frag-ments assize standards(lanes M). (A) RNAwashybridizedtothe HhaIUL38-13-galactosidaseprobe describedin Materials and Meth-ods and in the legend to Fig. 3. The panel on the left shows an experimentwith the -48/+99 and the -280/+99 gC recombinant viruses. The panel on the right shows an experiment with the -48/+99 RL recombinant virus. (B) ThesameRNAsamples from the gC recombinant virus-infected cells used in panel A were hybridized with the dUTPase probe described in Materials and Methods and thelegend toFig.4.
investigated the minimal sequence elements required for
measurable UL38 promoter activity with recombinants in both gC and the RL regions. Reporter constructs with promoters extending to -29 bases expressed the same
levels of chimeric
UL38-p-galactosidase
mRNA as did those with -48 bases of promoter in both locations (Fig. 6A). A promoter construct extending to only -23 bases, however, expressed nomeasurable level of chimeric mRNA. This is shown for a gC recombinant in Fig. 6B. These results are somewhat at variance with previously reportedresults oftransient-expression assaysinwhich the -23 promoter still had about30% of theactivityof themoreextensive ones (6). Extremely long exposures of similar RNaseprotection gelsandexaminationofhybridsformedby using more than twice the amount of infected-cell RNA revealedno RNaseprotection productcorrespondingtothe chimeric
UL38-p-galactosidase
mRNA(datanotshown).On the basis of controlexperiments with mixtures ofchimeric andwt mRNAs, this places levels ofexpression from the -23 promoter constructs as being lower than 5% of wtlevels.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.68.301.77.230.2]AC
A.
wt M PgC Recomb.
8.
UL38(G-gal
I1
+ +
C) a)
N. N.3M
c'J M
UL38/
-k-go'
-:
q8
-
J838/)S-ga
- 138() o)
c) r()
N~ C'.m
RL Recomb.
[image:7.612.64.291.74.386.2]gCRecomb.
FIG. 6. UL38promoteractivity requiressequencesextendingno
furtherthan 29 bases 5' of thetranscript capsite. Total RNAwas
isolated 8 h p.i. from rabbit skin fibroblasts and hybridizedto the
HhaIUL38-,-galactosidaseprobedescribed inMaterials and
Meth-ods and the legend to Fig. 3. After treatment with RNase, the protected fragmentswerefractionated on an8% denaturing
acryl-amide gel with HaeIII-digested pBR322 DNA fragments as size
standards(M). (A)The leftpanelshows results ofRNaseprotection
with 10 p,g of RNA from cells infected with wt, -48/+99, and
-29/+99 gCrecombinant viruses.Thispanelalso includesalane in
which600,000cpm(Cerenkov)ofundigested probewasloaded(lane
P). The full-length probe was 197 bp because it included pGEM polylinkersequences. The panelonthe rightshowsthe results of using10p.gof total RNAfrom cells infectedwith wt,-48/+99,and -29/+99RLrecombinant viruses.(B)RNaseprotectionwith 10pg
of total RNA isolated from twogCrecombinantviruses, -29/+99
and-23/+99.
DISCUSSION
Oneof thereasonsforinitiatingtheexperimentsdescribed in this communication was to develop an internally con-trolled system in which the expression ofHSV promoters bearingdefined modifications could be measured and
com-paredwith that of the unmodifiedparentalpromoter from the sameviralgenome. Itisimportanttoemphasize that
differ-ences in expression do not reflect some sort ofnonspecific
interference due to transcription through the inserted
re-portergene-promoter construct since all RNase protection and Northern blot analyses confirm the lack of detectable levels ofsuch readthrough RNA. Such a system is clearly feasibleon the basis of the datapresented here,which also demonstrate that theactivity ofamodel promoter
control-lingthesametranscriptcanvarybymorethan afactor of 5 depending on where it is expressed on the viral genome.
Sucharesult haspractical significance in the design of HSV vectors for moving and expressing specific genes into spe-cific infected tissuesorcells. It is also important in interpret-ing the results of deletional and substitutional mutagenesis studieson HSV promoter function and in the identification of promoter sequences controlling the kinetics of expression ofparticularHSV genes.
Ourpicture of the essential sequence elements in -y
pro-moters is generally based on the modifications of the gC promoter in situby Homa et al. (11-13). These researchers havedemonstrated thataverylimited amount of sequence 5' of the capsite, aswellas some elements 3', of it are all that
are required for full activity. Further, these workers have suggestedthat the sequenceof the TATA box element itself (-28to -23 of thecap) isadetermining factor in its activity (11-13).Similar in situ modification analyses on other model -y promoters such as that controlling the expression of mRNAs encoding the gH (UL22),
-Y42
(UL49),
andU.11
proteins generally confirm the limited extent of the 5' se-quencerequiredtocontrol strict latetranscription, although it is clear that no one specific TATA box sequence is conserved (16, 17, 22, 24). The present data on the UL38 promoter, which removes the influence of any possible sequence elements extending farther upstream than the regionsmodified in such in situ promoter modification stud-ies, fully support this general picture of HSV y promoters and demonstrate that very limited amounts of sequence elements upstream of thetranscription cap, along with other potentialelements 3' of thissite, provideall theinformation necessary for the expression oftranscripts following viral DNAreplication (6).The dataontheextentof theUL38 promoter upstream of the cap sitepresentedhereareingeneralagreementwithour transient-expression data presented earlier (6), with the notableexceptionthat in the viral genome theregion extend-ing between -29 and -23 corresponding to the sequence TaITAAAisabsolutelyessential for promoterfunction; thus, the canonical sequence TATAoccurring at -17of the cap site cannot alone mediate any appreciable transcriptional activity.This result is incontrastwith the situation found in transient-expression assays, in which constructs extending to -27 and -23 of the cap site (the latter eliminating the whole upstream T+A-rich element) had activities on the order of30% of full promoterexpression.Such dataprovide agood demonstration ofthe limitations of the use of tran-sient-expressionassaysas asole measureof promoter activ-ityinmutational modificationanalyses.
Although all available evidence demonstrates that the 5' extentof HSVy promotersisquite limited and is transport-ableas cassettes todifferent locations of the viral genome, it isclear that the environment of thiscorepromotercanhave a significant role in the levels of its activity. In the UL38 promoter, the addition ofarelativelysmallamount ofextra
sequencecorrespondingtotheUL37promoter and upstream sequencesin their normal
position
relativetoUL38
make they promoter
significantly
more active in thegC region used forgeneratingrecombinants.Two observations suggest that the low level ofactivityof the
minimally
activeUL38promoter ingC
isnot duetothe general inabilityofapromotertobeefficiently expressed
in this region. First, inclusion ofextra sequencesignificantly
increasesactivity.
Second,experiments currently
inprog-ress in which 219 bases ofthe dUTPase promoter is
being
usedtodrive theexpression
of similarchimericP-galactosi-dase-containing
constructs demonstrateessentially
equiva-lent levels of mRNAexpression
compared
with the wton November 9, 2019 by guest
http://jvi.asm.org/
mRNA. It isnot
clear, however,
whether the lowactivityofthe minimal
UL38
promoter is due to its kinetic class or whether minimal promoter elements of other classes oftranscripts
would also be expressed at lower than wt levelsin this region.
AlthoughthecoreUL38promoteris sufficienttodefine the
kinetic class of this gene, its
significantly
lower activity inthe
gC
location thanin theRLimpliesthat this minimal corepromoter does not contain all the information requirements for normal levels of mRNA
expression.
Although DNAsequence elements inserted upstream of the core UL38 promoter in the
gC
region are important in influencing itslevel of
expression, they
donotappeartobehighlyspecific. This conclusion is based on the fact that the same corepromoter
placed
inthe RLregion
inanorientation inwhichthe promoter
replaces
220 bases of the LAT promoter isactive at levels
approaching
those seen for the promoter inits normal location. Further, the same level of activity is
seenwhenthewholecassetteisinverted in the RLregionso
that the sequences
immediately
5' of the UL38corepromoter arecompletely
different. Thus, three different sets of up-stream sequences, onlyone of whichencodes anotherpro-moter-regulatory region, permit
essentially full activity of thecore promoter.Other data supportourconclusion that the local environ-ment of the promoter, as well as the specific sequences within
it,
isimportant
ininfluencing
levels ofexpression of HSVtranscripts.
Roemeret al.(20)
found that the location ofan inserted SV40promoter-enhancer-controlled reporter gene in the viral genome affected the levels ofexpression.Further,
removal ofrecognizable
ao4-bindingsites within the promoter of the L TK(UL23)
transcript has a profoundactivity
upon itsexpression
in transient-expression assays, but substitutionofsuchmodified promoters for thewtonein the viral genomedoesnotleadtoloss ofTKexpression (15). Asimilar observation has beenreportedforgD(23, 25).This result has beeninterpreted
as indicating that the viral ge-nomehasahigh
degreeofredundancyin sequence elements which bind either cellular or viral transcription factors sothat loss ofone or a few in anygiven region canbereadily
compensated
for. Our data would suggest that the DNA sequence upstream of the core UL38 promoter which in-cludes theUL37
promoteracting
in the opposite direction and the DNA sequence in the RL regions in which this promoter was recombined contain enough such sites that UL38 promoteractivity
ishigh.
However, our data also show that such sitesare not uniformlydistributedthrough-out the viral genome since the region within the gC locus used forrecombination isa
significantly
poorerenvironment fortranscription
than is theRLregion.ACKNOWLEDGMENTS
This work was supported byU.S. Public HealthService grants CA11861 and A106246. Further support was provided by the UC Irvine ResearchUnit in Animal Virology. J.F.G. isa predoctoral trainee and therecipientoftraininggrantT32GM07311 ("Structure andsynthesis ofbiologicalmacromolecules").
REFERENCES
1. Anderson, K.P., R.J. Frink, G. B. Devi-Rao, B. H. Gaylord, R. H.Costa,and E.K.Wagner.1981.Detailed characterization of the mRNAmappingintheHindIlI fragmentKregion of the herpessimplexvirus type 1 genome. J.Virol. 37:1011-1027. 2. Anderson, K. P., L. E. Holland, and E. K. Wagner. 1980.
Characterizationofherpessimplexvirus type 1 RNA present in theabsence of de novoprotein synthesis.J.Virol. 34:9-27. 3. Blair, E. D., C. C. Blair, and E. K. Wagner. 1987. Herpes
simplexvirusvirion stimulatoryproteinmRNAleader contains sequenceelements which increase bothvirus-induced transcrip-tion and mRNA stability. J.Virol. 61:2499-2508.
4. Dobson, A. T., F. Sedarati, G. B. Devi-Rao, W. M. Flanagan, M. J. Farrell, J.G. Stevens, E. K. Wagner, and L. T. Feldman. 1989. Identification of the latency-associated transcript pro-moter by expression of rabbit beta-globin mRNA in mouse sensory nerve ganglia latently infected with a recombinant herpes simplex virus. J. Virol.63:3844-3851.
5. Draper, K. G.,G. B. Devi-Rao, R. H.Costa,E. D.Blair,R. L. Thompson, and E. K. Wagner. 1986. Characterization of the genesencodingherpes simplexvirus type 1 and type 2alkaline exonucleases andoverlappingproteins.J. Virol. 57:1023-1036. 6. Flanagan,W.M.,A. G.Papavassiliou,M.Rice,L. B.Hecht, S. Silverstein, and E. K. Wagner. 1991. Analysis of the herpes simplex virus type 1 promoter controlling the expression of UL38, a true late geneinvolved in capsid assembly. J. Virol. 65:769-786.
7. Flanagan, W. M., and E. K. Wagner. 1987. A bi-functional reporter plasmid forthe simultaneous transient expression
as-say of two herpessimplex viruspromoters. Virus Genes 1:61-71.
8. Frink,R. J., R.Eisenberg,G.Cohen,and E.K. Wagner. 1983. Detailed analysisof the portionofthe herpessimplex virus type 1 genomeencodingglycoprotein C.J. Virol.45:634-647. 9. Gilman,M. 1989. Preparation and analysisofRNA, p.
4.7.1-4.7.8. In F. Ausubel, R. Brent, R. Kingston, J. Moore, J. Seidman, J. Smith, and K. Struchl (ed.), Current protocols in molecular biology.Wiley-Interscience, NewYork.
10. Holland, L. E., K. P. Anderson, C. Shipman, and E. K.Wagner. 1980. Viral DNAsynthesis isrequired for the efficient expres-sion of specific herpes simplex virus type 1 mRNAspecies. Virology 101:10-24.
11. Homa, F. L., J. C. Glorioso, and M. Levine. 1988. Aspecific 15-bp TATA boxpromoterelement is requiredfor expressionof aherpes simplexvirustype 1 lategene. Genes Dev. 2:40-53. 12. Homa, F. L., A. Krikos,J. C.Glorioso, and M. Levine. 1991.
Functional analysisof regulatoryregionscontrolling strict late HSV gene expression, p. 207-222. In E. K. Wagner (ed.), Herpesvirus transcription and its regulation. CRC Press, Inc., Boca Raton,Fla.
13. Homa, F. L., T. M. Otal, J. C. Glorioso, and M.Levine. 1986. Transcriptional control signals of a herpes simplex virus type1 late (gamma2) gene lie within bases -34 to + 124 relative to the 5' terminus of the mRNA. Mol.Cell. Biol. 6:3652-3666. 14. Honess, R. W., and B. Roizman. 1974. Regulation of herpesvirus
macromolecular synthesis. I. Cascade regulation of the synthe-sis of three groups of viral proteins. J. Virol. 14:8-19. 15. Imbalzano, A. N., A. A. Shepard, and N. A. DeLuca. 1990.
Functional relevance of specific interactions between herpes simplex virus type 1ICP4 and sequencesfrom the promoter-regulatory domain of the viral thymidine kinase gene. J. Virol. 64:2620-2631.
16. Johnson, P. A., and R. D. Everett. 1986. The control ofherpes simplex virus type-1 late gene transcription: a 'TATA-box'/cap site region is sufficient for fully efficient regulated activity. Nucleic Acids Res. 14:8247-8264.
17. Johnson, P. A., C.MacLean,H. S. Marsden, R. G. Dalziel,and R.D. Everett.1986. The product of gene
U.11
of herpes simplex virus type 1 is expressed as a true late gene. J. Gen. Virol. 67:871-883.18. Maniatis, T., E. F. Fritsch, and J. Sambroolk 1982. Molecular cloning: alaboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
19. McGeoch, D. J. 1989. The genomes of the humanherpesviruses: contents, relationships, and evolution. Annu. Rev. Microbiol. 43:235-265.
20. Roemer, K., P. A. Johnson, and T.Friedmann.1991. Activityof the simian virus 40 earlypromoter-enhancer in herpessimplex virus type 1vectors is dependent on its position, the infected cell type, and the presence ofVmwl75. J. Virol. 65:6900-6912. 21. Roizman, B., and A. E. Sears. 1990. Herpes simplex viruses and theirreplication, p. 1795-1842. In B. N.Fields, D. M. Knipe,
on November 9, 2019 by guest
http://jvi.asm.org/
R. M. Chanock, M. S. Hirsch, J. L. Melnick, T. P. Monath, and B. Roizman(ed.), Virology, 2nd ed. Raven Press, New York. 22. Shapira, M., F. L. Homa, J. C. Glorioso, and M. Levine. 1987.
Regulation of the herpes simplex virus type 1 late (gamma 2) glycoprotein Cgene:sequencesbetween base pairs -34to+29 control transientexpression and responsiveness to transactiva-tion by the products of the immediate early (alpha) 4 and 0
genes.NucleicAcids Res. 15:3097-3111.
23. Smiley, J. R.,D.C. Johnson, L.I.Pizer, and R. D. Everett. 1992.
The ICP4 binding sites in the herpes simplex virus type 1 glycoprotein D(gD)promoterarenotessential forefficient gD transcriptionduring virus infection. J. Virol. 66:623-631. 24. Steffy, K. R., and J. P. Weir. 1991. Upstreampromoterelements
oftheherpes simplex virustype1glycoprotein Hgene.J. Virol. 65:972-975.
25. Tedder,D. G., R. D. Everett, K. W. Wilcox, P. Beard, and L.I.
Pizer. 1989. ICP4-binding sites in the promoter and coding regions of the herpes simplex virus gD gene contribute to
activationofin vitrotranscription by ICP4. J. Virol. 63:2510-2520.
26. Wagner, E. K. 1985. Individual HSV transcripts:
characteriza-tion of specific genes, p. 45-104. In B. Roizman (ed.), The herpesviruses,vol. III.Plenum Press,NewYork.
27. Wagner, E. K., G. B. Devi-Rao, L. T. Feldman, A. T.Dobson, Y. F.Zhang, W. M. Flanagan, and J. G. Stevens. 1988.Physical characterization of the herpes simplex viruslatency-associated transcriptin neurons.J. Virol.62:1194-1202.
28. Wagner, E. K., W. M. Flanagan, G. B. Devi-Rao, Y. F.Zhang, J. M. Hill, K. P. Anderson, and J. G. Stevens. 1988. Theherpes simplexviruslatency-associatedtranscript is spliced during the latentphase of infection. J. Virol. 62:4577-4585.
29. Weinheimer, S. P., and S. L. McKnight. 1987. Transcriptional andpost-transcriptional controls establish the cascade ofherpes simplexvirusprotein synthesis.J. Mol.Biol. 195:819-833. 30. Zhang, Y. F., G. B. Devi-Rao, M. Rice, R. M. Sandri-Goldin,
andE. K.Wagner. 1987. The effect of elevated levels ofherpes simplex virus alpha-geneproductsonthe expression of model early and lategenesin vivo. Virology157:99-106.
31. Zhang, Y. F., and E. K. Wagner. 1987. The kinetics of
expres-sion ofindividual herpes simplex virustype1transcripts. Virus Genes1:49-60.