Polyomavirus Late Leader Region Serves
an
Essential Spacer
Function Necessary for
Viability and Late Gene Expression
GUY R. ADAMIAND GORDON G. CARMICHAEL*
DepartmentofMicrobiology, University of Connecticut Health Center, Farmington, Connecticut06032 Received 23 September1985/Accepted 20 January 1986
Allthreepolyomaviruslate mRNAs contain multiple tandem copies of thesamenontranslated57-nucleotide sequence,the lateleader,attheir5'ends. We show here thatapolyomavariant (ALM) lacking 48central bases ofthe 57-base leader unit is nonviable by plaque assayand by a newmethod fortesting virus viability, an immunofluorescence burstassay.ALM is, however, unaffected in early geneexpressionasmeasured both by indirect immunofluorescence of large T antigen and by transformation levels ofratF- 11lcells. DNA replication inmousecellsisalsoaswild type,andthe defect in ALM iscomplemented byanearly-defectivehelper virus DNA. ALM does not make detectable levels of late viralproteinsand isminimally 200-fold depressed in the accumulation ofcytoplasmic polyadenylatedlate RNA. When thedeleted leadersequenceof ALM isreplaced byavariety ofprocaryoticsequences, viabilityalmostalwaysreturns.Some of the substituted leader variants produceplaques with thesameapparentkineticsaswild-type viral DNA. The indication is that the sequence ofthepolyoma late leader is not important for lategeneexpression but that it hasanessentialspacerfunction on the RNA orDNA level. This spacer function is apparently necessary for late viral RNA transcription, processing, orstability.
The 5'-nontranslated sequences of polyomavirus late mRNAs are unusually heterogeneous. At least 15 different capped termini map throughout a 125-nucleotide region, from nucleotide5075 tonucleotide5168 on the viral genome
(5, 12, 17, 42). In addition, late messages contain variable
numbersofa reiterated 57-nucleotide late leader sequence
(nucleotides 5020 to 5076) at their 5' ends (24, 41). The
polyomavirus genome is circular, and owing to inefficient transcriptional termination, RNA polymerase II can make
multiple circuits while transcribinginthe lateorientation(1,
2). The result is giant
primary
transcripts consisting oftandemgenomicrepeats.
Thelate leader sequence (present in a single copy in the DNA) is part of the 400 base pairs (bp) making up the nontranslated regulatory region of the polyomavirus ge-nome.Figure1Adiagrams this region ofthe DNA. This area
ofthe genome has been much studied as it contains the
replication origin, the early transcriptional enhancer, and promoters for both earlyand latetranscription (5, 7, 8, 18,
27, 32, 43, 44).Bordering both ends ofthe lateleader unitare
excellent splice donor and acceptor sites. The model for processing ofmultiple-genomic-length transcripts is
repre-sented in Fig. 1B for a two-genome-length transcript. By intramolecular splicing all but the 57-base late leader se-quence is spliced out as a genome-length intron. The net result is that cytoplasmic mRNAs coding for late viral
proteinshave multiple5' leaders. The late leader unit itself has not beenexaminedin muchdetail.Within the late leader sequence there is a 10-nucleotide sequence that shows
strong complementarity tothe 3' end ofmouse 18S rRNA (38). Nuclease protection studies have revealed 40S ribo-somalsubunit-bindingsitesspecificallywithin the late leader sequence (23). These findings support a model in which multiple late leaders promote ribosome binding and thus moreefficient translation of late messages.
Beyond thepreliminary investigation mentioned above it
*Corresponding author.
is not known why the virus uses such aseeminglyinefficient
method to generate multiple late leaders. In an effort to
determine leader function we created a group ofmutants with most of the late leader deleted and replaced with
unrelatedsequencesof somewhat similar length. Ourresults
withthese mutantsdemonstrated the necessityofthe leader sequencefor virus
viability,
butapparently onlyas aspacer sequence. Bases 5026 to 5073(48 ofthe 57basesofthelateleader) are expendable but must be replaced by another
sequenceforthe virus to be viable.
MATERIALS ANDMETHODS
Materials.Enzymeswerefrom NewEnglandBioLabs,Inc. (Beverly, Mass.). a-32P-labeled deoxynucleoside triphos-phates (800Ci/mmol)werefromNewEngland Nuclear Corp.
(Boston, Mass.) or AmershamCorp. (Arlington Heights, Ill.). Fluorescein-conjugated goat antirat and rabbit antigoat antibodies were from Cooper Biomedical, Inc. (West
Chester,
Pa.).Synthetic oligonucleotides,
used asprimers
forsequence verification ofmutants, were synthesized with a
Systec model 1450A synthesizer. Hybond-mAP-paper for
RNAisolationwas fromAmersham.
Cells and culturetechniques. Cells weregrown on plastic
dishes with DulbeccomodifiedEagle medium
containing
5%(NIH 3T3) or 10% (F-111) calfserum and antibiotics. Pas-sagewascarried outwithatrypsin
(0.025%,
wt/vol)-EDTA
(0.2%, wt/vol)
solution prepared in phosphate-bufferedsa-line(PBS). NIH3T3 mouse cells and
F-ill
ratcellswere asdescribedpreviously(13).
F-li
cells wereused fortransfor-mation studies as described previously (13), and NIH 3T3 cellswereusedforplaqueassays(13),propagationofmutant viruses, and immunofluorescence studies.
Mutant construction. The starting vectorfor mutant con-struction was plasmid pPy, which is the entire genome of
polyomavirusstrain59RA(11,37)insertedattheBamHI site into pBR322. This plasmid was propagated in Escherichia
coli GM48 (F+ lac gal ara thrglu phi
T1,
T6r dam3 dcm6sup'),
obtained from Henryk Cudny, or in HB101. The417
on November 10, 2019 by guest
http://jvi.asm.org/
A
POLYOMA ORIGIN /PROMOTER REGIONreplication ori
m
100 1/5295
late stort sites
[image:2.612.130.484.267.372.2]AAA{
loteRNA... vv-vvv-vvlaeu_ leaderunit
5i00
enhoncer region 5100
rT W 1w l
it
BcHinfIBclI 5000
B
PROCESSING
OFTWO
GENOME-LENGTH
PRIMARYTRANSCRIPT
leader-to-leader splice leader-to-bodysplice
5 LL VPI body LL VPI AAAAAA
Mature VP I Message 5 LL LL* VPI body ' AAAAAA
FIG. 1. (A) Region of thepolyomavirusgenomewhich contains the early and late mRNAstartsites, the enhancer for early transcription, theorigin ofreplication, and the late leader unit. The stippledarearepresentstheregion of the leaderexonwhich has been deleted in the nonviable mutantALM.ori, Origin. (B) Model for processing of late primary transcripts and production of multiple, tandem nontranslated leaders. Polyomavirus late mRNAs contain variable numbers of tandem, 57-nucleotide nontranslated "leader" unitsattheir5'ends. These
aregenerated by splicing from giant primary transcripts resulting from inefficient transcription termination. The example shown is for a
primary transcript created bytwocircuits of RNA polymeraseIIaround theviralgenome.Thefinal mRNA containstwotandem leaderunits (LL) attachedtoa"body"segmentencoding the major structural protein, VP1. For all latemessages,the firstinitiator AUG codon is within
themessagebodysegment.
abbreviated leader mutant, ALM, was formed by deleting most of the sequences from the unique Bcll site at
polyomavirus nucleotide 5021 (numbering system asin
ref-erence 38) to the Hinfl site at nucleotide 5076 in pPy. Plasmid pPy DNA was linearized with BclI, and the ends were made blunt with DNA polymerase I, Klenowenzyme
(28). These molecules were then treated with calf intestine alkalinephosphatase followed by digestionwithBglI. Sepa-rately, theHinfl fragment spanning nucleotides 5073to 385
was isolated from pPy, the ends were made flush with the
Klenowenzyme, and then thefragment was cutwith BglI. Although several BglI sites arepresentinthevector, itwas
possibletotakeadvantageof the fact thatBglItendstoleave unique sticky ends. WhentheBglI-digested Hinfl fragment
wasgel isolated and then ligatedtotheBclI-BglI-cutvector, theresultwas acomplete pPylackingaBclI-Hinfl fragment. This construct (ALM) carriesa48-bp deletionin the leader exonbutretainsauniqueBcll site and 3bp junctionaltothe leader-leader spliceacceptor siteatthe 5' end of the leader
exonand6bp adjacenttothesplice donor siteatthe3'end of the leader. As for all mutants constructed, the entire sequencefrom theBclI siteatnucleotide5021totheBglIsite at nucleotide90 wasverified by subcloning into phage M13 vectorsfollowed by dideoxy DNA sequencing.
SLM M, SLM MR, and SLM B were constructed as follows. Plasmid pBR322 was digested withBglI andRsaI.
The 365-bpfragment spanning nucleotides 3482to 3847was gel isolated and partially digested with Sau3AI. The result-ant fragments were ligated into BclI-cut, phosphatase-treated ALM DNA. SLM M contains an insert ofpBR322
sequences 3689 to 3736 in the leader region. SLM MR contains the same sequences, but in opposite orientation. SLM B contains thesameinsertasSLMM,but also has 18 additional basesas aresult ofonly partial Sau3AI cleavage of thepBR322 BglI-RsaI fragment.
To construct SLM C, phage M13mp8 replicative form DNAwas digested withBglII and ClaI, and the endswere made flush with the Klenow enzyme and then ligated to BclI-cut, filled-in ALM molecules. Unexpectedly, the final clone from this procedure also contained an insert of M13mp8sequencesfrom nucleotides 6935to 6884.
SLM MP8 and SLM MP8R containessentiallythecloning region from M13mp8, in opposite orientations. M13mp8 replicative form DNA was digested withBamHI, the ends were made flush with Klenow enzyme, andthen the mole-cules were religated. This was done to destroy theBamHI site, as future transfection experiments require a single BamHI site in the polyomavirus genome. These molecules wereusedtogeneratewhiteplaqueson alawnofJM103cells
(31) (phage M13mp8 gives blue plaques under the same
conditions), demonstrating the +1 frameshift in the lacZ coding region. Thesephageswerethen propagated,andRF 200
eorly RNA
4900
3'
3'
on November 10, 2019 by guest
http://jvi.asm.org/
molecules were isolated and cut with HindIlI and EcoRI,
and the ends made flush with Klenow enzyme. The small
fragmentwasligated into BclI-cut, phosphatase blunt-ended ALM DNA.
Plaque assays. Plaque assays (13) were done in 35-mm
petri dishes seeded 24 h before infection with 105 3T3 cells.
Cells were transfected with 0.1 to 1 ,ug of ligated
BamHI-digested recombinant plasmid DNA by the DEAE-dextran
method (29) followed by an agar overlay. Wild-type and mutant (ALM or SLMs) DNAs were used in these assays.
Dishes were examined for plaques from 5 to 12 days after
transfection.
Immunofluorescence. Immunofluorescence assays were donewithNIH 3T3 or 3T6 cells which had been transfected with 0.1 to 0.5 ,ug of mutant or wild-type DNAs by the
calcium phosphate technique (16) as modified by Wigler et al. (46)andwith aglycerol shock(33). Alternatively, similar resultswere obtainedwhentransfections were done by the DEAE-dextran method (29) immediately after which the
cells were shocked for 1 min with 20% glycerol in HEPES
(N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic
acid)-buffered saline. One day before transfection cells were
platedat 2 x 105/60-mmdish. Dishes contained glass cover
slips. The DNAs used were first digested with BamHI to
release the viralgenome from pBR322 sequences and then
recircularized withDNAligase to reconstruct intact, infec-tious polyomavirus genomes. At the times indicated
posttransfection, coverslipswereremoved andrinsedonce
with PBS lacking calcium andmagnesium (PBS-; 144 mM
NaCl, 2.7 mM KCl, 15 mM sodium phosphate, pH 7.4).
They were then fixed for 5 min at -20°C with methanol-acetone (1:3), drained, andallowed to dry. They were then washed once with 5% goat serum in PBS- followed by an
H20
wash. Stainingwithprimary antibodywas donefor 30 min at23°C. Next, two washes with 5% goat serum in PBS-weredonefollowed by two washes with PBS-. Fluorescein-conjugated secondary antibody was added and allowed toincubate for 30 min at 23°C. Finally, slides were washed three times with PBS-, once with H20, and then mounted with glycerol and examined under a fluorescence micro-scope.
The burst assay basically followed the procedure for
immunofluorescence assayswithNIH3T3cells. Cells were
plated1daybeforeoncoverslips,at4 x
105
cells per60-mmplate.At30 hposttransfection, onecover slip was removed and stainedforlarge Tantigenas apreburstcontrol.At 3 and 5days staining for largeT wasdone to detect bursts. It was
helpful to include a wild-type transfection to control for
density of cells which might affectburst size.
Analysis of RNA levels. Each 90-mm culture dish, seeded with 8x
i05
NIH3T3cells 24 h before, wastransfectedwith 15 ,ugof BamHI-digested, recircularized plasmid DNA by thecalcium phosphate coprecipitation method(16) asmod-ifiedby Wigleret al. (46) and followed by a glycerol shock
(33). Cytoplasmic RNA was isolated by the method of
Favaloro et al. (9) except that polyadenylated RNA was
further purified by affinity chromatography with poly(U)
covalently bound to diazo-thiophenyl paper obtained from Amersham(47). Thisprocedureshould also remove all small DNAoligonucleotides producedby DNase I treatment at an earlier step in the purification. Polyadenylated RNA from one90-mmplateof cells waspurifiedwith 1 cm2ofpoly(U) paper as instructedby the manufacturer. Carrier yeast tRNA
(20
ptg/ml)
was addedtothepolyadenylated RNA in water,andthis was ethanol precipitatedandstored at -20°C until use. One-tenth of each polyadenylated RNA sample was
suspended in 0.5
,ll
of 25 mM NaH2PO4, pH 7.2. This wastransferred to GeneScreen Plus (New England Nuclear Corp.) with a hybri-slot manifold slot blot apparatus (Be-thesda Research Laboratories, Gaithersburg, Md.). Filters were treated for hybridization as described by Church and Gilbert (3). DNAprobes used in hybridization were prepared
essentially as described by Feinberg and Vogelstein (10). DNA was cleaved withApaI, BamHI, and EcoRI.
Electro-phoresiswas done in a 1% low-melting-temperature agarose minigel in Trisacetate buffer with 1 ,ug of ethidium bromide per,ul. A 1,371-bp fragmentcorresponding topolyomavirus nucleotides4632 to3261 (late probe) and a 1,051-bp fragment
(nucleotides 1560 to 2611) (early probe) were excised from the gel. H20was then added at a ratio of 3 ml of H20 per g of gel, and the samples were boiled for 7 min to melt the agarose and denature the DNA fragments. Labeling with
[ci-32P]dATP
and random-sequence synthetic hexadeoxy-nucleotides (made with a Systec model 1450 DNA synthe-sizer and thephosphoramidite chemistry) were asdescribed previously (10). Quantitation of RNA was done by densito-metric scanning of the slot-blotautoradiograms.RESULTS
Construction and analysis of abbreviated leader mutant ALM. As a first step in examination of the function of the late leader, we deleted the sequences between the BcII site
(nucleotide 5026) and the Hinfl site (nucleotide 5073) in a
polyomavirus-pBR322 recombinant molecule. This
con-structionthuscarriesadeletion of 48 of the original 57 bases of the leader exon and yields a viral genome with a 9-base leader sequence (Fig. 1A). This abbreviated leader mutant (ALM)maintainsauniqueBclI site and 3 bpjunctionalto the
leader-leadersplice acceptor site at the 5' end of the leader exon and 6 bpadjacentto the splicedonor site at the 3' end of the leader. Thelocalizationof thedefect in the mutant was
confirmed by cloning a wild-type BclI-BgIl fragment into ALM which restored viability. Also, the sequence of this
fragment from ALM was verified by subcloning into
bacteriophage M13mp8DNAfollowed bydideoxy
sequenc-ing.
To test for viability, we transfected NIH 3T3 cells with
BamHI-liberatedALMgenomesfrom therecombinant plas-mid or with wild-type polyomavirus genomes similarly cut from recombinant molecules. In repeated attempts ALM
failed to produce plaques in a plaque assay, while cloned wild-type polyomavirus DNA consistently gave 50 to
60
plaques per 100 ng in the same assay. ALM was next
examined to determine whether early viral functions were normal. Indirectimmunofluorescence was used todetermine whether NIH 3T3 cells transfected with BamHI-linearized
ALM DNA produced intranuclear large T antigen. The
results areshown inFig. 2. Similar levelsofpositive nuclei
were observed with ALM, and pPy, indicating that large T antigen is produce.d in the abbreviated leader mutant. As another measure of early gene expression (this time middle T
antigen), the entire ALM plasmid was used to transform Fisher ratF-111 cells. Results were similar to those with the wild-type (59RA) control plasmid, pPy (Table 1). DNA replication assays (data not shown) indicated that the ALM
BamHI-linearized DNA was replicatedto the samelevel as pPy
BamHI-linearized
DNA at 30 h posttransfection. To demonstrate conclusively that the defect in ALM is not in early gene expression or replication, we performed mixed DNAtransfections with ALM and acloned early-defectivehelper DNA(lacking the
XbaI
fragment,nucleotides2477 toon November 10, 2019 by guest
http://jvi.asm.org/
30
hrs
anti-T
30
anti-VP1
hrs
PPY
[image:4.612.63.557.76.421.2]ALM
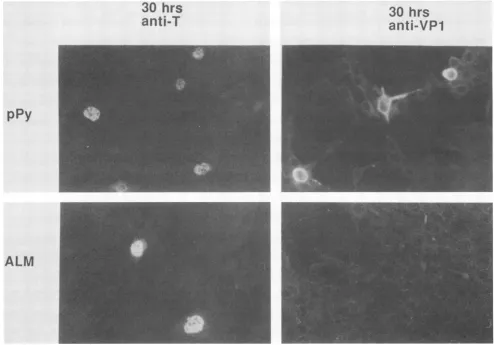
FIG. 2. Immunofluorescence localization of earlyandlate viralproteinsafterDNAtransfections.PlasmidDNAs pPy(wildtype)andALM
werecutwithBamHI,and polyomavirus genomes wererecircularized withDNAligaseandtransfectedby the DEAE-dextranmethod into
mouse3T3cells. At 30h aftertransfection, coverslipswerefixed andreacted with eitherratanti-Tserum(early)orrabbitanti-VP1serum
(late).Fluorescein-labeled secondary antibodywasusedtoallowvisualizationof viralantigens. 2522; unable to produce fully functional large T antigen).
Thismixedtransfectiongaveplaquescontainingamixture of
the complementing defective genomes. This mixed virus stock yielded plaques in 6to 7days,asdidwild-type virus. These plaques were picked and amplified, and the virus stockswereused forproductive infections. After isolation of
low-molecular-weight DNA and restriction enzyme diges-tion, it was seen that the ALM and helper genomes were presentinapproximately equalamounts(datanotshown). It should be noted that this finding eliminates the possibility that ALM is nonviable because the deleted sequence con-tains aDNA-packagingsignal.
Indirectimmunofluorescencewithantiseraspecific forlate proteins was next done to determine whether late gene expressionwas normal in the mutantlate after DNA trans-fection. AlthoughNIH3T3 cells transfected withwild-type
cloned DNA gave up to 5% positive nuclear fluorescence withanti-VP1serum,nopositiveswere even seenwith ALM
(Fig. 2). Resultsweresimilar whenacombined antiserum for
theothertwoviral lateproteins, VP2 and VP3, was used. RNAanalysis was done to evaluatefurther the defectin ALM. Aslotblot ofpolyadenylated cytoplasmic RNA from ALMandpPy viral DNA-transfected cellswasdone(Fig. 3).
For theseblots 5to10timesmoreALM RNA thanwild-type
RNAwas applied to the slot-blot apparatus to show more
clearly the nature of the ALM defect. ALM has been shown to be indistinguishable from wild-type viral DNA in early gene expression and DNA replication after DNA transfec-tion. For this reasonhybridization of RNA to an internally labeledprobe specificforearlyRNA wasused to control for transfection efficiency and RNA recovery from the transfected cells. An identical blot ofpPy and ALM
poly-adenylated cytoplasmicRNA washybridizedto aninternally labeled probe specific for late RNA. Thus, although the
signal of early polyomavirus RNA from ALM-transfected
TABLE 1. Transformation ofF-111 cellsbypPy and ALM'
Plasmid AmtofDNA(jig) No.offoci
pPy 0.1 35,24,35
1.0 38,53, 48,60
ALM 0.1 15,25, 20,30
1.0 43, 58, 65, 41
Control 0 0,0
a106cellsper90-mmdishweretransfected withtheindicatedquantitiesof
plasmid DNA,asdescribed in Materials and Methods. Fociwerecounted at 14days.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.320.561.617.698.2]Cytoplasmic
poly)pPy
9 1
Early Probe
Late Probe
.i
I.
i..
FIG. 3. Slot-blot analysis of early and latecyt viral RNA after transfection of NIH 3T3 cellsw
DNA. RNA was preparedas described inMateri and applied to the slot-blot manifold in two d (relativeamounts,9 and1).Thefilterswerehybrid late specific32P-labeledprobes andsubjectedto a cells was considerably more intense than f same samples the opposite was true when
probed (Fig. 3). Densitometric scanning of
vealed that minimally there is a 200-fold red polyadenylated late RNA accumulated in th
transfected cells.
Analysis of substituted leader mutants ('
spliced correctly the leader inALMwouldb
A
Wild Type (59RA)
.AAG(AGUCGGGAGGAAALMACUGUGUUGGAGGCCCtICcGCCAUUCGcUGAUCAA)GUA.
Abbreviated Leader Mutant(ALM)
..AAG(AGU...deletlonof nt 5026-5073.*-GAUCAA)GUA..
+ ninenucleotides)thatSImapping to examineleadersplicing
A~
mRNA would probably not work. Therefore, in an effort to con-struct a variant that would be easier to study, severalsubstituted leader mutants(SLMs) were created by inserting ALM variousshort
procaryotic
sequencesinto theuniqueIclI
siteof ALM. This restored nearly correct length leaders and 9 would also be agood control for the change in spacing of regulatory elements in the DNA of the deletion
mutant
& & ALM. The leader sequences ofsome of these mutants are
3 s shown inFig. 4. To oursurprise,manyofthesewere viable, twoof whichgave plaqueson NIH 3T3 cellswith the same
kinetics aswild-type pPy DNA.
Alternate method to determine virusviability. Anempirical findingis that when NIH 3T3 cells aretransfected with viral
DNA and stained at 48 hfor large T antigen, occasionally
toplasmic polyA+ dense clusters of 60 to 100 positive nuclei are observed.
iith pPy or ALM These have been assumed to be viral bursts which would ials and Methods occurwhen transfected cells lyseproducing virus that
sec-lifferent amounts ondarily infects surrounding cells. When staining isdone at
lized
withearly
or 3 and 5 days posttransfection, virtually all that is seen isLutoradiography. bursts. This method was used to test the substituted leader
mutantsforviabilityasit wasfaster than the standardplaque
or pPy, for the assay and
potentially
more sensitive. Indirectimmunofluo-late RNA was rescenceforlarge T wasdoneat3 and also 5daysso as not such blots re- tomiss variantsthatmightbe slowgrowing. Figure5 shows
luction in ALM results from atypical burst assaydone here with wild-type
ie cytoplasm of pPy and with ALM. At day 1 bright
positive
nuclei were presentfor bothDNAs. Atday5pPy-transfected cellswere SLMs). If it is positive for bursts whiletheALM-transfected cells showedFe soshort(only no evidence of virus
production
and reinfection. Thissup-B
VIRUS
LEADER
LENGTH
59RA (WT)
--It
SLM/MP8 (M13mp8 cloning region inserted)
..AAG(AGUGAUCAGCtUGGCUGCAGGUCGACGGAUCGAUCCCCGGGAAUUGAUCAA)GUA..
SLM/MP8R (Ml3mp8 cloning region,other orientation)
..AAG(AGUGAUCAAUUCCCGGGGAUCGAUCCGUCGACCUGCAGCCAAGCUGAUCAA)GUA_
SLM Y(yeastspacer insert)
..AAG(AGUGAUCUGACAAGCGUUUGCGAAGCUCCUGAAUCAAUAAGAAGGUGAUCAA)GUA-SLMC(Ml3mp8spacer insert)
..AAG(AGUGAUCCGUUUUUAUUUUCAUCGUAGGAAUCAUGAUCUACAAAGGCUAUCAGGUCAUUG
CCUGAGAGUCUGGAGCAAACAAGAGAAUCGGAUCAA)GUA..
SLM B (pBR322 spacer insert, nt3736-3671)
-AAG(AGUGAUCGGAGGACCGAAGGAGCUAACCGC ACAACAUGGGGGAUCAUGUAAC UCGCCUUGAUCAA)GUA..
SLMM (pBR322 spacer insert,nt 3736-3689)
..AAG(AGUGAUCGGAGGACCGAAGGAGCUAACCGCUUUUUUGCACAACAUGGGGGAUCAA)GUA.
SLM/MP8 MlSm0 cloning reion
SLM/MP8R l-bl13mps cloning region \
SLMY
-N
yeastepcw insrEn]
SLM MR (pBR322 spacer insert, nt 3689-3736)
..AAG(AGUGAUCCCCCAUGUUGUGCAAAAAAGCGGUUAGCUCCUUCGGUCCUCCGAUCAA)GUIA SU1 C -Li
AUG M13mp spacerInsert
SLMB -N pBR322 AUG AUG KNt
FIG. 4. Mutants in the late leader. (A)The top line shows the sequence of the wild-type leader unit along with several flanking bases.ALMwasconstructedasdescribed in MaterialsandMethods bydeleting sequencesbetweentheBclI siteatnucleotide (nt)5021 SLM M and theHinflsiteatnucleotide5076.Thesubstitutedleadermutants
wereconstructed byinsertingDNAfragmentsinto the unique
Bc(I
siteretainedbyALM.(B) Schematicrepresentation ofthe mutants. AUG codons areindicated. WT, wildtype. SLMMR-IN pBR322
73 nt
55 nt
AUG
s\ N ALM57 nt
9 nt
51 nt
51 nt
52nt
9l\\I-6
ntpBR322 &\ 55 nt
----H.AUQ
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.57.297.76.220.2] [image:5.612.59.568.391.728.2]30
HRS
120
HRS
PPY
ALM
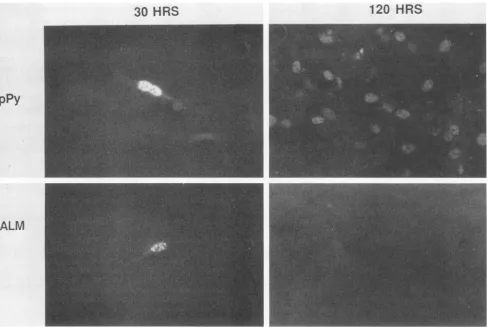
FIG. 5. Burst assay to test ALMforviability. Plasmid DNAs frompPy(wildtype) and ALM were cutwithBamHI,andpolyomavirus genomes wererecircularized withDNAligaseandtransfectedbytheDEAE-dextranmethodintomouse3T3cells.Atdays1and 5coverslips werefixedandstained with rat anti-T serumfollowed byfluorescein-labeled secondaryantibody.
ports the plaque assay finding that ALM is nonviable.
Evidently
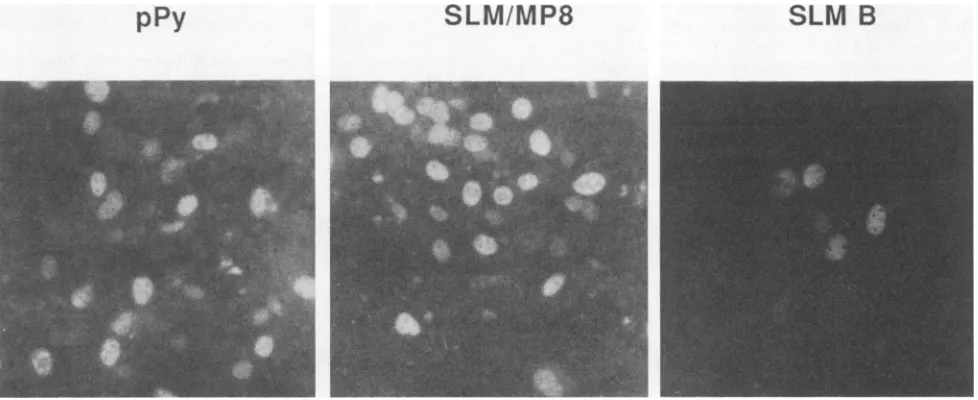
by day5transient expression of largeTislargely undetectable forALM (and presumably pPy). Burst assays were alsoused to test substituted leader mutants for viabil-ity. Figure 6shows examples ofbursts
for pPy, SLM MP8,and SLM B. Note
that
there is an inverse correlation between sizeofburstandlength of timerequired to produceplaquesinplaque assays (Table 2). Because of the small size
ofthebursts with someof the mutantstrains, itishelpfulto
transfect withlow levelsofDNA.That way, even at3days,
groups of four to six positives are definitely due to viral burstsand notchance clusters of cellstransientlyexpressing large T antigen. Note that all constructions that did not produce plaques were also negativein the burst assay.
DISCUSSION
We constructed and studied a group of polyomavirus mutants with altered late leader sequences. The 9-base
abbreviatedleadervariant, ALM, isnonviable and doesnot produce detectable late proteins or detectable cytoplasmic late mRNA, indicatingthat the leader is necessary for late gene expression. Restoration of viability (and late gene
expression) by insertion of arbitrary sequences into the ALM abbreviated leader suggests thatamajor leader
func-tionisasaspacer (onthe DNA or RNAlevel). Toreiterate, the spatial setup of the leader region is important but the actualleader sequence is not, at least for the 48 bases deleted in ALM.
Early speculations on polyomavirus late leader function
(23, 24, 41) and numerous studies of mRNA 5' leaders in general have proposed a role for leaders in control of mRNA translation. One example is in adenovirus type 5: the dele-tion of the 5' tripartite late leader results in a fivefold decrease in translation efficiency ofarecombinant message (26). The polyomavirus wild-type late leader has apurported ribosome-binding site (23), no internal AUGs, and aregion
complementary to the 3' end ofmouse 18S rRNA (38). A translational control model for polyomavirus late messages would suggest that the leader sequence promotesribosome binding and thus late mRNA translation. Tandem leaders would potentiate this effect. Ouroriginal finding that ALM was nonviable supports this model, butourlater results do not.
When growth characteristics of the substituted leader mutants were examined, it became apparent that the nature of thelate leader could indeed affect late geneexpression but generally not in any unexpected manner. SLM M and SLM MR create leaders ofnearwild-typelength. Both,however, containAUG codons with no in-frame termination codons in the new leaders. Normally, the5'-most AUGs in eucaryotic mRNAs are used for translational initiation. Studies have shown(22, 25) that if an additional AUG is inserted 5' to the original start codon, inhibition of translation from the down-streamAUG results. In SLM M and SLM MR this would be expected to cause the formation of a late translational frameshift and lower levels ofcorrectly initiated wild-type
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.70.557.71.400.2]Ppy
SLM/MP8
I
SLM B
FIG. 6. Characteristic bursts for wild type andtwosubstituted leadermutants.Methodswere asdescribedin thelegend forFig. 6, but staining was doneat 3days after transfection with the DNAs.
polypeptides. Not all of these AUGs have surrounding
sequences which obey the consensus for translational
initi-ation (21). However, since they will presumablybe present
multiple times in most late mRNAs ofthese viruses, any negative effect on translation will be multiplied.
It has been demonstrated (22, 25) that when an in-frame
termination codon is inserted after an "artificial" 5'-most
AUG, translation again occurs at the original downstream start codon atclose to wild-type levels. Sequence datafor SLMBandSLMC inFig.5reveal thatthismaybethecase
forthe leadersinthese variants.Asonemight predict, SLM
B and SLM C are both viable but produce plaques more
slowlythan thewildtypedoes.Itshouldbe noted thatSLM
B has twoleader AUGs within 9bases of each other, with only one having an in-frame terminator. This somewhat
complicates ourexplanation forviability ofthis mutant.
Finally, SLM MP8 and SLM MP8R(withthesameleader
insertbutintheopposite orientation)have nointernalleader
AUGs,aleaderlength of51bases,andproduce plaqueswith the same kinetics as wild type. These, along with SLM Y, are the three examples of AUG codon-free substituted leaders that were tested. Whether theidentity ofthe leader sequence,outsideof being AUG-free,is irrelevant togrowth
in tissue culture requires the study of more substituted
TABLE 2. Plaque assays oflateleadermutants"
Virus Time of appearance of
plaques (days)
pPy (WT)... 6-7 ALM... Nonviable
SLM MP8...7...67 SLM MP8R... 6-7 SLMY ... 6.5-7.5 SLM C ... 9-10 SLMB ... 9-10 SLMM... Nonviable
SLM MR... Nonviable
leadermutants.If the polyomaleader sequencedoeshavea rolein translational controlit maybesubtle or not sequence
specific.The 48bases deletedinALM removethepurported
ribosome-protected region (23) and the 10 bases which are somewhat complementary to the 3' end of 18S rRNA (38). These arereplaced withother sequences in theviable SLMs.
Our results do notsupporta primarytranslational defect in ALM.Rather,ALMisalmostcertainly defective because of
its deficiency inlate cytoplasmic mRNA. Late mRNAwith
an abbreviated leader may, however, additionally be trans-lated poorly. One might speculate that mRNA which is translated poorlyis less stable inthecytoplasm, but results with two typesofadenovirusmutantsaffected in late mRNA
translation do not show this (26, 40).
Simian virus 40 (SV40), another papovavirus, resembles
polyomavirusin itsmode of late geneexpression.A common 5' leader is found in some form on all late mRNAs. Also similar is theheterogeneity ofthe 5'endsoflate mRNAs and the dependency on large T antigen for late transcription
initiation. It isdifficult, however, to apply resultsfrom SV40 to polyomavirus. Unlike polyomavirus, the late leader in
SV40 mRNA is over 240 bases long, is not repeated, contains internal splice sites, and codes for a protein, the
agnoprotein (14, 15, 20, 30, 36). A large numberof viable
leader deletion mutants of SV40 have been reported (36). Some shifting upstreamoflate mRNA startsites occurs in some ofthese mutants, and many lack internal splice sites (34, 35). Somasekhar and Mertz (39) have recently shown
thatbydeletingoneSV40 late leaderintron splicedonor site orbymakingan insertionin the same area, the patternof5' RNAstartsiteschanges.Thefrequencyofsplicingatsplice
donor and acceptor sites hundreds of bases away is also affected.
Althoughthere isnoprecedencefordownstream promoter elementsin SV40 late genes, ourresultsareconsistent with the leader functioning on the DNA level as a necessary spacer between two promoter elements. Onecan speculate
that without the leader region present late transcriptional
initiation could be inhibited oraltered. This might resultin decreased steady-state levels ofALM cytoplasmic mRNA
owingto decreased initiationorincreased degradation.
aPlaque assays were done on NIH 3T3 cells with DNAs excised from
recombinant plasmids and recircularized with DNAligase asdescribed in Materials and Methods.WT,Wildtype.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.66.555.76.276.2]Anothermodel for the ALM defect is that the lesion is in RNA splicing, resulting in an inability to form mature cytoplasmic mRNA molecules. The late leader in polyoma-virus may simply function as a spacer to allow efficient splicing for removal of genome-length introns. In ALM the leader contains 9 bases between the leader splice acceptor siteand the leadersplice donor site. The deletion is in the exon; intron sequences and splice junctions are intact. Unlikeintrons, which showconsensus sequences and mini-mal sizesfor function (80bases in a rabbit,B-globinmessage)
(45), exon requirements for splicing are not well defined. Maybe splicingcannot occur in thissystembecause strong splice donor and acceptor sites are so close together. It is
possible that a short exon would inhibit proper splicing
owing to steric hindrance ofthe splicing machinery. If in ALMtheleader-to-leader splicesandleader-to-bodysplices for mVP1 andmVP3 occur at very lowlevels, nonviability of
this virus would be explained. Increasingthe leaderlengthin ALM to 51bases (SLMMP8) and to even greater lengths in otherconstructions might create an exon larger than what may be a minimum length requirement for splicing in this system.
At this time it is not clear whether the primary defect in ALMisat the levelofRNAsynthesis,processing, transport, or degradation. An examination of late RNA produced by mutant ALM is under way to determine rates of RNA
synthesis(not steady-state levels) and whether RNAsplicing
is occurring efficiently. These studies should reveal more
clearlythe natureof the spacer function of thepolyomavirus
late leader.
Finally,avirus relatedtopolyomavirus hasbeen isolated fromhamsters(6). It will be of interest to learn whether the hamsterpapovavirus also directsthe synthesis of giantlate
nuclear RNAsand tandem late leader units.
ACKNOWLEDGMENTS
WethankHermanWolf forhelp withsomeofthefiguresandJanice Seagren for typingthemanuscript.
Thisworkwassupported by Public Health ServicegrantCA32325 from the NationalCancerInstitute,agrant-in-aid from the American HeartAssociation, andagrantfromtheAmerican CancerSociety. G.C. isanEstablishedInvestigator oftheAmericanHeart Associ-ation. Theoligonucleotide synthesizerwaspurchased withmatching funds fromtheDepartment of Defense.
LITERATURE CITED
1. Acheson,N. 1978.PolyomagiantRNAscontain tandem repeats of the nucleotide sequence of the entire viral genome. Proc. Natl. Acad. Sci. USA 75:4754-4758.
2. Acheson, N. 1976. Transcription during productive infection withpolyoma virusandsimian virus40.Cell8:1-8.
3. Church, G.,and W.Gilbert. 1984. Genomicsequencing.Proc. Natl. Acad. Sci. USA 81:1991-1995.
4. Cowie, A.,C. Tyndall, andR. Kamen. 1981. Sequencesatthe capped5'-ends ofpolyomavirus lateregionmRNAs:an
exam-ple of extreme terminal heterogeneity. Nucleic Acids Res. 10:6305-6322.
5. Dailey,L., and C. Basilico. 1985.Sequencesin thepolyomavirus DNA regulatory region involved in viral DNAreplication and earlygeneexpression. J. Virol. 54:739-749.
6. Delmas, V.,C. Bastien,S.Scherneck,andJ.Feunteun. 1985.A new member of the polyomavirus family: the hamster papovavirus. Completenucleotide sequence andtransformation properties. EMBOJ. 4:1279-1286.
7. deVilliers, J., L. Olson, C. Tyndall, and W. Schaffner. 1982. Transcriptional "enhancers" from SV40 and polyoma virus showacell typepreference. NucleicAcids Res. 10:7965-7976. 8. deVilliers, J., and W. Schaffner. 1981. A small segment of
polyoma virus DNA enhances the expression of a cloned
P-globin
gene over adistance of 1400 base pairs. Nucleic AcidsRes. 9:6251-6264.
9. Favaloro, J., R. Treisman, and R. Kamen. 1980.Transcription maps of polyoma virus specific RNA: analysis by two-dimensional nuclease S1 gel mapping. Methods Enzymol. 65:718-749.
10. Feinberg, P. F., and B. Vogelstein. 1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 132:6-13.
11. Feunteun, J., L. Sompayrac, M. Fluck, and T. L. Benjamin. 1976. Localization of gene functions in polyoma virus DNA. Proc. Natl. Acad. Sci. USA 68:283-288.
12. Flavell, A. J., A. Cowie,S. Legon, and R. Kamen. 1979.Multiple 5'-terminal cap structures in late polyoma virus RNA. Cell 16:357-372.
13. Fluck, M., and T. L.Benjamin.1979. Comparison oftwoearly gene functions essential for transformation in polyoma virusand SV40.Virology 96:205-228.
14. Ghosh, P., V. Reddy, J. Swinscoe, P. Choudry, P. Lebowitz, and S. Weissman. 1978. The5'-terminal leader sequence of late 16S mRNAfrom cells infected with simianvirus40. J.Biol. Chem. 253:3643-3647.
15. Ghosh, P., V. Reddy, J. Swinscoe, P. Lebowitz, and S. Weis-sman.1978.Heterogeneityand 5'-terminal structures of the late RNAs of simianvirus 40. J. Mol. Biol. 126:813-846.
16. Graham, F. L., and A. J. van der Eb. 1973. A new techniquefor the assay ofinfectivity ofhumanadenovirus5 DNA. Virology 52:456-467.
17. Heiser, W. C., and W. Eckhart. 1982. Polyomavirusearlyand latemRNAsinproductively infectedmouse3T6cells.J.Virol. 44:175-188.
18. Herbomel, P., B. Bourachot, and M. Yaniv. 1984. Two distinct enhancers with different cellspecificitiescoexist in the regula-toryregion of polyoma. Cell39:653-662.
19. Hirt, B. 1969. Selective extraction of polyoma DNA from infectedmouse cell cultures.J. Mol. Biol.26:365-369. 20. Jay, G., S. Nomura, C. W. Anderson, and G. Khoury. 1981.
Identification of the SV40 agnogene product: a DNA binding protein. Nature(London)291:346-349.
21. Kozak, M. 1981. Possiblerole offlanking nucleotides in recog-nition of the AUG codon by eukaryotic ribosomes. Nucleic AcidsRes. 9:5233-5252.
22. Kozak, M. 1984. Selection of initiation sites by eucaryotic ribosomes: effect ofinserting AUG triplets upstreamfrom the coding sequence for preproinsulin. Nucleic Acids Res. 12:3873-3893.
23. Legon, S. 1979. Binding of ribosomestopolyoma virus RNA. Possibleroleof the leaderregion ininitiation siterecognition.J. Mol. Biol. 134:219-240.
24. Legon,S.,A.Flavell, A. Cowie, and R. Kamen. 197. Amplifica-tionin the leader sequenceof latepolyoma virusmRNAs.Cell 16:373-378.
25. Liu,C.-C.,C. C.Simonsen,and A. D. Levinson.1984.Initiation of translation at internal AUG codon in mammalian cells. Nature(London)309:82-85.
26. Logan, J., and T. Shenk. 1984. Adenovirus tripartite leader sequenceenhances translation of mRNAs late after infection. Proc.Natl. Acad. Sci. USA 81:3655-3659.
27. Luthman, H., M. G. Nilsson, andG. Magnusson. 1983. Non-contiguoussegments of thepolyomagenomerequired in cisfor DNAreplication.J. Mol. Biol. 161:533-550.
28. Maniatis, T., E. Fritsch, and J. Sambrook. 1982. Molecular cloning,alaboratory manual. ColdSpringHarborLaboratory, ColdSpring Harbor, N.Y.
29. McCutchan, J., and J. Pagano. 1968. Enhancement of the infectivity of simian virus 40 deoxyribonucleic acid with diethylaminoethyl-dextran. J. Natl. CancerInst.41:351-356. 30. Mertz, J., A.Murphy,and A.Barkan.1983. Mutantsdeleted in
the agnogene of simian virus 40definea newcomplementation group. J. Virol. 45:36-46.
31. Messing, J. 1983. New M13 vectors for cloning. Methods
on November 10, 2019 by guest
http://jvi.asm.org/
Enzymol. 101:20-78.
32. Muller, W. J., C. R. Mueller, A. M. Mes, and J. A. Hassell. 1983. Polyomavirus originfor DNA replication comprises mul-tiple genetic elements. J. Virol. 47:586-599.
33. Parker, G., and G. Stark. 1979. Regulation of simian virus 40 transcription: sensitive analysis of the RNA species present earlyin infections by virusorviralDNA. J. Virol. 31:360-369. 34. Piatak, M., P. K. Ghosh, L. C. Norkin, and S. M. Weissman. 1983. Sequences locating the5' endsof the major simian virus 40latemRNAforms.J. Virol.48:503-520.
35. Piatak, M., K. N. Subramanian, P. Roy, and S. M. Weissman. 1981. Late messenger RNA production by viable simian virus 40 mutants with deletions in the leader region. J. Mol. Biol. 153:589-618.
36. Reddy, V., P. Ghosh, P. Lebowitz, and S. Weissman. 1978.Gaps and duplicated sequences in the leaders of SV40 16S RNA. NucleicAcids Res. 5:4195-4213.
37. Ruley, E., and M. Fried. 1983. Sequence repeats in a polyomavirus DNA region important for gene expression. J. Virol.47:233-237.
38. Soeda,E., J. Arrand, N.Smolar, J. Walsh, and B. Griffin. 1980. Coding potential and regulatory signals of the polyomavirus genome.Nature(London)283:445-453.
39. Somasekhar, M., and J. Mertz. 1985. Exon mutants thataffect the choice of splice sites used in processing the SV40 late transcripts. Nucleic AcidsRes. 13:5591-5609.
40. Thimmappaya, B., C. Weinberger, R. J. Schneider, and T.
Shenk. 1982. Adenovirus VAI RNA is required for efficient translation of viral mRNAs at late times after infection. Cell 31:543-551.
41. Treisman, R. 1980. Characterization ofpolyoma late mRNA leader sequences by molecular cloning and DNA sequence analysis. NucleicAcidsRes. 8:4867-4888.
42. Treisman, R., and R. Kamen. 1981. Structure of polyoma virus late nuclear RNA. J.Mol. Biol. 148:273-301.
43. Tyndall, C., G. LaMantia, C. Thacker, and R. Kamen. 1981. A region ofthe polyoma virus genome between the replication origin and late protein coding sequences is required in cis for both early gene expression and viral DNA replication. Nucleic AcidsRes. 9:6231-6250.
44. Veldman, G., S. Lupton, and R. Kamen. 1985. Polyomavirus enhancer contains multiple redundant sequence elements that activate both DNA replication and gene expression. Mol. Cell. Biol.5:649-658.
45. Wieringa, B., E. Hofer, and C. Weissman. 1984. A minimal intronlength but no specific internal sequence is requiredfor splicingthelarge rabbit 1-globin intron. Cell 37:915-925. 46. Wigler, M., A. Pellicer, S.Silverstein,R.Axel, G.Urlab, and L.
Chasin. 1979.DNA-mediated transfer oftheadenine phospho-ribosyltransferase locus into mammalian cells. Proc. Natl. Acad. Sci. USA 76:1373-1376.
47. Wreschner, D. H., and M. Herzberg. 1984. A new blotting medium for the simple isolation and identification of highly resolved messenger RNA. Nucleic Acids Res. 12:1349-1359.