Pimobendan B from powder diffraction
data
Alvis Zvirgzdins,a* Mara Delina,aAnatoly Mishnevb and Andris Actinsa
aUniversity of Latvia, Department of Chemistry, Kr. Valdemara Street 48, Riga,
LV-1013, Latvia, andbLatvian Institute of Organic Synthesis, Aizkraukles Street 21,
Riga, LV-1006, Latvia
Correspondence e-mail: [email protected]
Received 15 September 2013; accepted 15 October 2013
Key indicators: powder X-ray study;T= 293 K; mean(C–C) = 0.003 A˚;Rfactor = 0.019;wRfactor = 0.026; data-to-parameter ratio = 49.6.
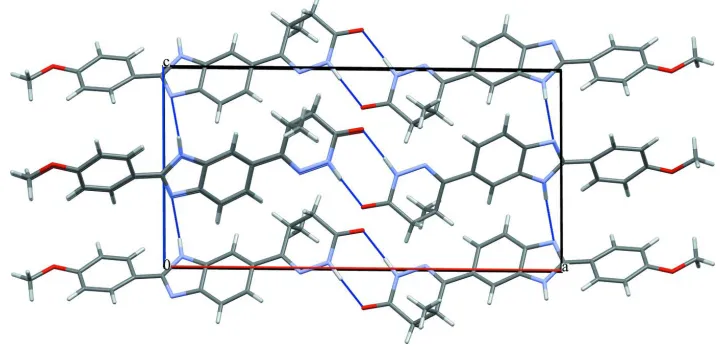
The title molecule, C19H18N4O2{systematic name: (RS )-6-[2-(4-methoxyphenyl)-1H -benzimidazol-5-yl]-5-methyl-4,5-di-hydropyridazin-3(2H)-one}, adopts an extended conforma-tion. The dihedral angles between the central benzimidazole ring sytem and the pendant methoxyphenyl and pyridazinone residues are 1.41 (18) and 9.7 (3), respectively. In the crystal, N—H N hydrogen bonds link the imadazole groups into [001] chains, and pairs of N—H O hydrogen bonds link the pyridazinone groups into dimers. Together, these generate a two-dimensional supramolecular structure parallel to (010). The layers are linked by C—H interactions.
Related literature
For general information about pimobendan, see: Gordonet al.
(2006). For related crystalline forms, see: Boerenet al.(2011). Semi-empirical calculations were carried out with
HYPERCHEM Professional(Hypercube, 2010). Refinement of lattice parameters and peak profile determination were performed by Le Bail profile fitting (Le Bailet al., 1988)
Experimental
Crystal data
C19H18N4O2 Mr= 334.37
Monoclinic,P21=c a= 18.891 (5) A˚
c= 9.5029 (8) A˚
= 90.799 (13)
V= 1788.2 (5) A˚3 Z= 4
= 1.54184 A˚
= 0.68 mm1 T= 293 K
cylinder, 160.5 mm
Data collection
Bruker D8 diffractometer Specimen mounting: capillary Data collection mode: transmission
Scan method: step 2min= 3.5
, 2max= 70.00
, 2step= 0.01
Refinement
Rp= 0.019 Rwp= 0.026 Rexp= 0.020 RBragg= 0.015
2 = 1.690
6651 data points 134 parameters 56 restraints
[image:1.610.313.564.297.346.2]H-atom parameters not refined
Table 1
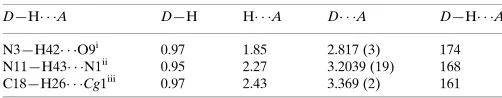
Hydrogen-bond geometry (A˚ ,).
Cg1 is the centroid of the C12/C20/C15/C24/C22/C21 ring.
D—H A D—H H A D A D—H A
N3—H42 O9i
0.97 1.85 2.817 (3) 174
N11—H43 N1ii
0.95 2.27 3.2039 (19) 168
C18—H26 Cg1iii 0.97 2.43 3.369 (2) 161
Symmetry codes: (i)xþ1;yþ1;z; (ii)x;yþ3 2;zþ
1
2; (iii)x;y 1 2;z
1 2.
Data collection:Dicvol(Boultif & Loue¨r, 2004); cell refinement:
FOX (Favre-Nicolin & Cˇ erny´, 2002); data reduction: FOX; program(s) used to solve structure:FOX; program(s) used to refine structure: FULLPROF (Rodriguez-Carvajal, 1993), CRYSTALS
(Betteridge et al., 2003) and PLATON (Spek, 2009); molecular graphics:Mercury (Macrae et al., 2008); software used to prepare material for publication:WinPlotr(Roisnel & Rodriguez-Carvajal, 2000) andpublCIF(Westrip, 2010).
This work was supported by the European Regional Development Fund (No. 2011/0014/2DP/2.1.1.1.0/10/APIA/ VIAA/092).
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB7139).
References
Betteridge, P. W., Carruthers, J. R., Cooper, R. I., Prout, K. & Watkin, D. J. (2003).J. Appl. Cryst.36, 1487.
Boeren, M. M. M., Paridaans, R. J., Petkune, S., Lusis, V. & Muceniece, Dz. (2011). US Patent No. 20,110,152,283 A1 20110623.
Boultif, A. & Loue¨r, D. (2004).J. Appl. Cryst.37, 724–731. Favre-Nicolin, V. & Cˇ erny´, R. (2002).J. Appl. Cryst.35, 734–743.
Gordon, S. G., Miller, M. V. & Saunders, A. B. (2006).J. Am. Anim. Hosp. Assoc.42, 90–93.
Hypercube (2010).HYPERCHEM Professional. Hypercube, Inc., Gainesville, Florida, USA.
Le Bail, A., Duroy, H. & Fourquet, J. L. (1988).Mater. Res. Bull.23, 447–452. Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008).J. Appl. Cryst.41, 466–470.
Rodriguez-Carvajal, J. (1993).Physica B,192, 55–69.
Roisnel, T. & Rodriguez-Carvajal, J. (2000).EPDIC,7, 118–123. Spek, A. L. (2009).Acta Cryst.D65, 148–155.
Westrip, S. P. (2010).J. Appl. Cryst.43, 920–925. Structure Reports
Online
supporting information
Acta Cryst. (2013). E69, o1677 [doi:10.1107/S1600536813028353]
Pimobendan B from powder diffraction data
Alvis Zvirgzdins, Mara Delina, Anatoly Mishnev and Andris Actins
S1. Experimental
Indexing of patterns was performed with WinPlotr (Roisnel & Rodriguez-Carvajal, 2000) and Dicvol (Boultif & Louër,
2004) using reflections in the 2θ range of 3.00 – 30.00°. Space groups for all polymorphs were determined using FOX
1.9.7.0 (Favre-Nicolin & Černý, 2002). The correct space group was selected based on possible systematic extinctions.
The compositions of unit cell and the values of Z were determined in all cases from the unit cell volume.
Refinement of lattice parameters and peak profile determination were performed by Le Bail profile fitting (Le Bail et
al., 1988) using FOX. Structures were determined with FOX by parallel tempering algorithm. The best cost function
values were reached by using automatic temperature schedule and Cauchy-type displacement amplitude schedule.
The input model of pimobendan molecule was obtained from semiempirical calculations by HYPERCHEM Professional
(Hypercube, 2010) for both - R and S enentiomer. The molecules were described in terms of Fenske-Hall Z-matrix format
ans structure solutions The dihedral angles C21—C22—O7—C25; N11—C8—C20—C15 and C2—C6—C14—N4 were
defined as intramolecular degrees of freedom and were varied during the structure determinations.
S1.1. Synthesis and crystallization
Pimobendan form B was prepared in three steps. At the first step, its dioxane solvate was held in a thermostat at 100°C
for one day. At the second step obtained powder were suspended in methanol and suspension were hold in a dry box
while all methanol evaporates. At the end obtained methanol solvate were desolvatated at 100°C.
S1.2. Refinement
Rietveld refinement for the final structure was performed by Fullprof. Hydrogen atoms were added with Crystals
according to the molecular geometry and their positions were not refined. Since the bond lengths and angles departed to
unacceptable values, atomic parameters for (N3, N4, C5, O9, C14, C16, C17,C23), (N1, N11, C2, C6, C8, C10, C13,
C18, C19) and (O7, C12, C15, C20, C21, C22, C24, C25) were refined as rigid bodies.
S2. Results and discussion
Several crysltalline forms of pimobendan and its preparation are patented (Boeren et al., 2011) but there are no crystal
data for these polymorphs or pseudopolymorhs. This article is focused on the structure determination from powder data
and description of the pimobendan B form.
Lowest value of cost function were obtained by using molecular model of R enantiomer in structure determination
process. The final structure of pimobendane B form shows that pimobendane molecule is almost linear because the
dihedral angle value of N11—C8—C20—C15 = 9.7 (3)° and C13—C6—C14—N4 = 1.41 (18)°. The crystal structure of
title compound consist of molecules that are conected via hydrogen bonds that are formed between two imidazole groups
molecules. Since pimobendan B form are obtained from its methanol solvate by desolvation at 100°C, these voids may be
result of desolvation at temperature that is almost twice as large as boiling point this solvent. Pimobendan B form at
ambient conditions tends to form monohydrate. Unstabilty of pimobendane B form at ambient conditions may be
[image:3.610.121.483.152.294.2]explained by penetration of water molecules into voids of crystal structure.
Figure 1
The molecular structure of the title compound showing 50% probability ellipsoids and hydrogen atoms are shown as
small spheres of arbitrary radii.
Figure 2
[image:3.610.124.484.349.522.2]Figure 3
Stacking interactions in the crystal structure of title compound
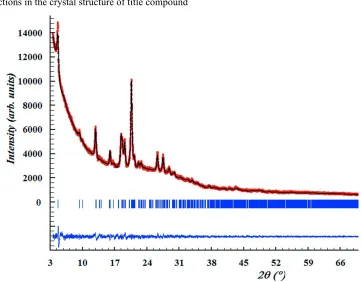
Figure 4
Scattered X-ray intensities of title compound at ambient conditions as a function of diffraction angle 2θ. The observed
pattern (red dots), the best Rietveld fit profiles (line) and the difference curve between the observed and calculated
profiles (below) are shown.
(RS)-6-[2-(4-Methoxyphenyl)-1H-benzimidazol-5-yl]-5-methyl-4,5-dihydropyridazin-3(2H)-one
Crystal data
C19H18N4O2
Mr = 334.37
Monoclinic, P21/c
Hall symbol: -P 2ybc a = 18.891 (5) Å
c = 9.5029 (8) Å β = 90.799 (13)° V = 1788.2 (5) Å3
Z = 4
[image:4.610.123.484.249.531.2]T = 293 K cylinder, 16 × 0.5 mm
Data collection
Bruker D8 diffractometer
Radiation source: sealed X-ray tube None monochromator
Specimen mounting: capillary Data collection mode: transmission Scan method: step
2θmin = 3.5°, 2θmax = 70.00°, 2θstep = 0.01°
Refinement
Refinement on Inet
Least-squares matrix: full Rp = 0.019
Rwp = 0.026
Rexp = 0.020
RBragg = 0.015
χ2 = 1.690
6651 data points
Profile function: Pseudo Voigt
134 parameters 56 restraints 75 constraints
Hydrogen site location: inferred from neighbouring sites
H-atom parameters not refined
(Δ/σ)max = 0.01
Background function: linear extrapolation
Special details
Refinement. Rietveld refinement for the final structure was performed by Fullprof. Hydrogen atoms were added with Crystals according to the molecular geometry and their positions were not refined, but final refinement was performed with hydrogen atoms. Since the bond lengths and angles departed to unacceptable values, atomic parameters for (N3, N4, C5, O9, C14, C16, C17,C23), (N1, N11, C2, C6, C8, C10, C13, C18, C19) and (O7, C12, C15, C20, C21, C22, C24, C25) were refined as rigid bodies.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C22 −0.21504 (17) 0.87352 (17) −0.02608 (17) 0.01267* C23 0.34340 (13) 0.78483 (13) 0.19844 (13) 0.01267* C24 −0.15891 (17) 0.93136 (17) 0.05152 (17) 0.01267* C25 −0.33907 (17) 0.86902 (17) −0.06608 (17) 0.01267* H26 0.13416 0.47697 −0.26987 0.0152* H27 0.24355 0.46651 −0.16258 0.0152* H28 0.18217 0.69913 0.16336 0.0152* H29 −0.05380 0.92376 0.09189 0.0152* H30 −0.16806 1.00299 0.11652 0.0152* H31 −0.24066 0.72624 −0.16210 0.0152* H32 −0.12618 0.63253 −0.17586 0.0152* H33 −0.38045 0.92304 −0.04037 0.0152* H34 −0.34437 0.78145 −0.02928 0.0152* H35 −0.33452 0.86891 −0.16468 0.0152* H36 0.27557 0.63704 0.27202 0.0152* H37 0.35319 0.45789 0.28346 0.0152* H38 0.39052 0.57826 0.36071 0.0152* H39 0.35524 0.82003 0.29000 0.0152* H40 0.30710 0.84168 0.15603 0.0152* H41 0.38474 0.78628 0.14061 0.0152* H42 0.44322 0.50470 −0.04847 0.0152* H43 0.03860 0.78640 0.13920 0.0152*
Geometric parameters (Å, º)
C22—O7—C25 116.05 (17) C14—C16—C17 108.38 (13) C8—N1—C19 107.21 (14) C14—C16—C23 107.98 (10) N4—N3—C5 123.69 (18) C17—C16—C23 111.36 (18) N3—N4—C14 118.38 (14) C5—C17—C16 109.74 (12) C8—N11—C10 106.35 (12) C13—C18—C19 115.67 (12) N4—N3—H42 107.00 N1—C19—C18 132.23 (13) C5—N3—H42 129.00 C10—C19—C18 121.47 (19) C10—N11—H43 127.00 N1—C19—C10 106.28 (15) C8—N11—H43 127.00 C8—C20—C12 122.36 (16) C6—C2—C10 120.30 (13) C8—C20—C15 119.7 (2) O9—C5—C17 129.25 (14) O7—C22—C21 127.2 (3) N3—C5—C17 115.3 (2) C10—C2—H28 121.00 O9—C5—N3 115.26 (18) C6—C13—H27 121.00 C13—C6—C14 118.42 (17) C15—C24—C22 120.41 (18) C2—C6—C13 118.65 (19) C12—C21—C22 119.6 (2) C2—C6—C14 121.70 (13) C21—C22—C24 119.3 (3) N11—C8—C20 125.48 (14) C6—C2—H28 119.00 N1—C8—C20 122.63 (16) C20—C12—H32 119.00 N1—C8—N11 111.60 (19) C20—C15—H29 119.00 N11—C10—C19 108.49 (17) C24—C15—H29 119.00 C2—C10—C19 120.71 (17) C21—C12—H32 118.00 N11—C10—C2 130.76 (12) C12—C20—C15 116.2 (3) C20—C12—C21 122.47 (18) C18—C13—H27 116.00 C6—C13—C18 123.12 (17) O7—C22—C24 113.20 (18) N4—C14—C6 115.89 (14) C14—C16—H36 110.00 C6—C14—C16 122.25 (17) C17—C16—H36 110.00 N4—C14—C16 121.6 (2) C23—C16—H36 109.00 C20—C15—C24 121.7 (2) C5—C17—H37 109.00
Hydrogen-bond geometry (Å, º)
Cg1 is the centroid of the C12/C20/C15/C24/C22/C21 ring.
D—H···A D—H H···A D···A D—H···A
N3—H42···O9i 0.97 1.85 2.817 (3) 174
N11—H43···N1ii 0.95 2.27 3.2039 (19) 168
C18—H26···Cg1iii 0.97 2.43 3.369 (2) 161