JOURNAL OF VIROLOGY, Sept. 1992, p. 5509-5515 0022-538X/92/095509-07$02.00/0
CopyrightX 1992, American Society forMicrobiology
A
Cell-Free
Recombination
System for Site-Specific
Integration
of
Multigenic Shuttle
Plasmids
into the
Herpes
Simplex Virus
Type
1 Genome
PHILIPJ. GAGE,' BRIANSAUER,2 MYRON LEVINE,3ANDJOSEPH C. GLORIOSO4* Department of Microbiology and Immunology, Universityof Michigan MedicalSchool, Ann Arbor, Michigan
48109-06201;
Experimental Station, E328, Dupont-Merck PharmaceuticalCompany, Wilmington,Delaware 19880-0328;DepartmentofHuman Genetics, University of Michigan MedicalSchool,
AnnArbor, Michigan 48109-06183; and Department of MolecularGenetics andBiochemistry
University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania152614
Received 23 April1992/Accepted 19 June 1992
Thisreport describesanovel method for complementation studies ofdefective herpes simplexvirus (HSV)
genes.Viraltestgeneand nonviral reportergenecassetteswererapidlyintegrated intotheHSVgenomeina
site-specific and reversible manner by using the P1 phage-based Cre-ox recombination system. Shuttle plasmids containedafunctionalloxP recombination site,anexpressible form ofthe bacterial lacZgene,anda
copyofthe
wild-type
glycoprotein B (gB) gene ordouble mutant gB allele containing both atemperature-sensitive (ts) mutation and a syncytium (syn)-forming mutation. A recipient viralgenome, KAT:dloxl, was
constructed from the HSV tpe 1 (syn) gB-deficient mutant virus, KAT, by marker transfer of the loxP
recombination site into the viral thymidine kinase locus. Shuttle plasmids ofup to 12.9 kb in length were
recombined with high efficiency(11to20%o)into theKAT::laxl genomein cell-free, Cre-mediated recombi-nation reactions. Expression ofa functional wild-type ordouble mutant gB polypeptide complementedthe nonfunctional polypeptide expressed from the deleted, normal gB locus and allowed production ofeither wild-type orSyn- plaques onVero cells. The latter recombinant viruswasalsotsfor growth. Theability to
express viral genes from plasmids which can be shuttled into and out of the HSV genome in cell-free
recombination reactions makes thisapowerfulmethodfor performinggenetic studies of thebiologicproperties of viralgeneproducts.
Herpes simplex virus type1 (HSV-1) belongstoafamily
of animal viruses typified by having large, complex DNA
genomes (100 to 250 kb) (for a review, see reference 25).
HSV-1 contains approximately 72 genes encodingproteins
which maybe classified as providing either essential
func-tions for infectious-particle production in cell culture or accessoryfunctions which contribute toviral pathogenesis and extend the virushostcellrangeduring natural infections
in vivo.Although the entire HSV genomicsequenceisnow
known (21-23), the biological properties and roles in repli-cation formany ofthese viral functions have notyetbeen elucidated.
Theproduction and characterizationof HSVmutantshave provided the central approachtouncoveringthe functional roles of individualgenes incarryingout differentphasesof thecomplexvirus lifecycle.Theprincipal strategyhas been to genetically alter cloned viral DNA segments and then study the affected biologicfeatures of the mutantproducts eitherintransient transfection experiments orin infections after marker transfer of the mutant gene into the viral
genomeby homologous recombination. While transient as-sayscanbe useful for the initial characterization ofmutant constructs,manyexperimental approaches requirethat mu-tantgenesbe transferred into the viralgenome (3, 7, 9, 11, 12, 29)foranalysisoftheirimpactincomplex settingssuch as infection of cell cultures or animals. Genetic studies whichrelyonmarker transferproceduresareusuallyslowed by the requirementforpriorcharacterization ofthe mutant
* Correspondingauthor.
constructand bya lowyield of recombinants. Asa
conse-quence, this approach is rather laborious, particularly for experiments requiring the analysis of large panels ofmutant constructs. Moreover, sequences transferred into the viral genome by homologous recombination are not directly
re-coverable butmustbe recloned priortofurther characteri-zation or manipulation. Amorerapid method for
introduc-tion of mutant constructs into the HSV genome, which would permit the identification of relevant mutations that resultinaparticular phenotype, would greatlyfacilitate the geneticandbiochemicalcharacterization of these viralgenes
and theirproducts.
Workby Saueretal.(28) demonstratedthatplasmidDNA couldbeefficientlyinserted intoaherpesvirusgenomeinan
in vitrorecombination reaction.By taking advantageof the Cre-lox site-specific recombination machinery of bacterio-phage P1 (2, 16, 33), a plasmid containing the 34-bp loxP recombination site, but completely lacking in homologous herpesvirus sequences, could be specifically inserted at a
loxP site engineered into the pseudorabies virus genome. Theinserted plasmid disruptedthe nonessential gIII glyco-protein gene. Recombination was efficient and could be accomplished in a cell-free reaction mixture consisting of onlythe viral andplasmidDNAsandthe Cre recombinase. Transfectionof the reactionmixture into suitableeukaryotic cells allowedfor therescueofinfectious recombinantvirus.
Recombinationefficiencies ofatleast8%werereported, and
the inserted plasmids were stably maintained in the viral
genome.Furthermore,because the Crerecombination
reac-tion is reversible, the inserted shuttle plasmid could be
recovered from the recombinant viral genome by a Cre-5509
Vol. 66, No. 9
on November 9, 2019 by guest
http://jvi.asm.org/
5510 GAGE ET AL.
mediated intramolecular recombination reaction and
by
propagation aftertransformation ofEscherichia coli.
Inthis report,wedescribehowthe Cre-lox shuttlevector strategy was adapted for complementation of an HSV-1
mutantwiththe essential glycoprotein B(gB)gene deleted.
Wild-typeand mutantcopies of thegBgene were
efficiently
shuttled into andout ofagB- loxP recipient virusgenome. Recombinant viruses were identified on the basis of their
blue-plaque phenotype derived from expression of a lacZ
reporter gene present within the inserted shuttle
plasmid.
Expression offunctional gB polypeptides from the inserted plasmids complemented the gB- defect of the
recipient
virus. Insertion of a temperature-sensitive (ts) and syn
double mutant gBalleleconferred bothmutant phenotypes
on therecombinant virus. These studies provided evidence that the Cre-loxshuttle systemwill beuseful for thegenetic
analysisof the rolesofboth viralgenesaswellas cis-acting genomicelements whichfunction during virusreplication.
MATERIALS AND METHODS
Cellsandvirusstrains. Verocells andthe D6cell linewere maintained in Eagle's minimum essential medium (MEM) (GIBCO Laboratories, Gaithersburg, Md.) supplemented
with 10 mM HEPES
(N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid), the nonessential amino acids, and 5% newborncalfserum (GIBCO Laboratories). D6 cells, which were previously derived by transforming Vero cells with
pKBXX (8) and pSV2neo (31), express HSV gB-1 upon infectionandcomplement the growth of gB- viruses (8). D6 cellsweremaintained inculturemedium supplementedwith 200jigof the antibioticG418(GIBCO Laboratories) perml. For transfection experiments, the cells were maintained in MEM supplemented with 5% fetal calf serum (GIBCO
Laboratories) instead of newborn calfserum.
KAT is a derivative of HSV-1 (strain KOS) carrying a
969-bp BstEII deletion within the essential gB gene and
expresses an internally deleted, nonfunctional gB
polypep-tide which is secreted into the medium (8). Both KAT and thecomplementingcellline, D6,werekindlyprovided by S. Person. KATandKAT::loxl (described below)were
propa-gated, andtheirtiters onD6 cellsweredeterminedat 370C. Cre-mediated recombinants derived from KAT::loxl were firstisolatedonD6cells and thengrown, and their titerson
Verocellsweredetermined at37°C. Recombinants contain-ing the ts form of gB were tested for this phenotype by plaqueassaysonVero cellsat34 and39°C. Allrecombinant virusstockswerederived fromplaque-purifiedisolates.
For plaque assays orindividual plaque isolations, serial dilutions of virus stocks were inoculated onto confluent monolayers of the indicated cell line which were
subse-quentlyincubatedunder methylcellulose (0.5% methylcellu-lose in MEM supplemented with 10 mM HEPES and 2% newborn calfserum) at 370C until plaques developed. The
cultures were then stained either with crystal violet (1%
crystal violetin 50% ethanol)orwithBluo-gal (halogenated
indolyl-13-D-galactoside) for ,B-galactosidase (,3-gal) activity
(see below), and when appropriate, virus from individual blue plaqueswasisolated for subsequentpurification.
Bacterialstrains.Plasmidswereisolatedandpropagated in
E. colistrainsDH5a (15)orGM119 (giftfromD. Oxender).
Plasmid-transformed bacteriaweregrowninLuria broth(10
goftryptoneperliter, 5gofyeast extractperliter, 171mM
NaCl) supplemented with 75 ,ug of ampicillin (Boehringer MannheimBiochemicals, Indianapolis, Ind.)permlor onL
agarplates containing ampicillin.
Construction of plasmids. All
cloning
steps andplasmid
propagations were
performed
by
standardprocedures
(15,
17,
27).
Enzymeswerepurchased
fromGIBCO-BRL,
Inc.,
or
Boehringer
Mannheim Biochemicals and usedasspecified
by the manufacturers.
pLink760
(34),
pON1
(32), pKBXX
(8), and pBS64 and pBS65
(28)
have all been described previously. pUCXI(19)
contains the HSV-1 BamHI Pfrag-ment
containing
thecomplete
thymidine
kinase(tk)
locus subcloned frompXl
(18)
intopUC9.
pJG101
wascon-structed from
pUCX1
by inserting
a354-bp
NaeI-SmaIfragment
containing
a functionalloxP
site frompBS64
intotheuniqueSnaBI site within the coding sequence for the tk gene. This354-bp insertionoccursbetween residues 110and
111 of the TK protein and was
expected
toabrogate
TKactivity. A 270-bp
BamHI-PvuII
fragment frompBS65,
includinga functional laxPsite, wasinserted into
pBSM13-(Stratagene,
LaJolla,
Calif.)
betweentheBamHI andSmaI sites, creatingpBSloxP. pIEPlacZ
was constructedby
sub-cloning a 763-bp
BglII
fragment from pLink760 into the uniqueBglII
site ofpON1,
placing a promoterless cassettecontaining the bacterial lacZ gene under the control ofthe human cytomegalovirus immediate-early promoter
(HCM
VIEP). The HCMVIEP-lacZ gene cassette from
pIEPlacZ
was inserted at the unique BamHI site of pBSloxP as a
4.7-kb BamHI fragment, creating
pJG103.
Finally, tocon-struct pJG108 (see Fig. 3),
pJG103
was partially digested withBamHI, the linear unit-length plasmidfragment
was gel purified, anda3.7-kb HSV-1BamHI-BclI
fragment contain-ing the entire gB-1 (strain KOS) gene from pKBXX was inserted in the orientation shown (see Fig. 3).pJG102
(see Fig. 3) was constructed in a similar fashion frompBSloxP by inserting (i) the ts syngB-1
allele from the HSV-1 mutant tsB5 asa 4.9-kbKpnI
fragment (0.346 to 0.377 map units) atthe pBSloxP
KpnI
site and (ii) the 4.7-kbBamHI
HCM VIEP-lacZ gene cassette from pIEPlacZ at the pBSloxP BamHI site.Viral DNA isolation. ViralDNA wasisolated by a modifi-cation of standard methods for high-molecular-weight eu-karyotic DNA isolation (4, 14). Confluent Vero cell
mono-layers were infected ata multiplicity of infection of 5. At 24 h postinfection, virions free in the medium and released by freeze-thawing cells were combined and pelleted at 75,000x g for 40 min. The virion pellet was resuspended in lysis
solution (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 0.6% sodium dodecyl sulfate, and 0.25 mg of proteinase K per ml)
and incubated at
37°C
for 8 to 12 h. DNA was multiply extracted first with phenol-chloroform (1:1) and then with chloroform, precipitated with isopropanol, and resuspended in TE (10 mMTris-HCl [pH 8.0], 1 mM EDTA) buffer.Marker transfer ofloxP into KAT. TheloxPsite in
pJG101
was marker transferred into the tk locus of the KAT genome by homologous recombination after cotransfection of viral
and plasmid DNAs into subconfluent D6 cells as described by Graham and van der Eb (13) and modified by Homa et al. (18). Because marker transfer of the loxP fragment would
interrupt TK expression, transfection lysates were enriched for TK- viruses (24, 26) by serial propagation at low
multiplicity of infection (<0.001) on D6 cells in MEM supplemented with 100
jig
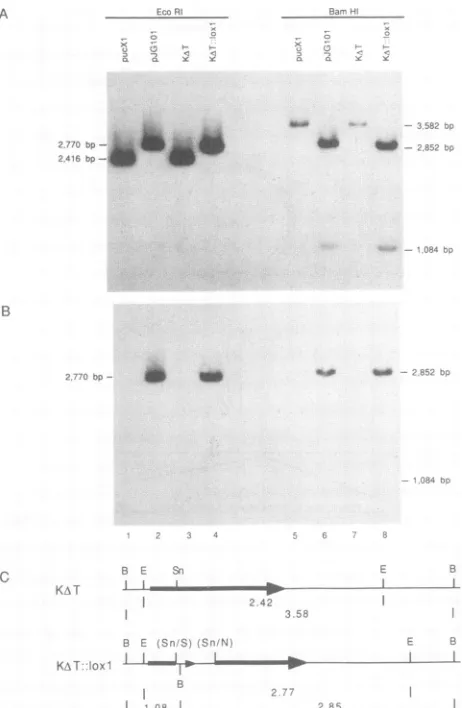
of thymine 1-,B-D-arabinofurano-side (araT) (Sigma Chemicals, St. Louis, Mo.) per ml. The enriched TK- stock was used to inoculate D6 cell monolay-ers, individual plaques were isolated, and small virus stocks were grown on D6 cells in microtiter plates. Infected cell lysates were tested for theloxPinsert by DNA dot blot assay with theNaeI-SmaI loxP fragment from pBS64 as probe.A recombinant virus,KAT::loxl,
containing theloxP
insert,J.VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
INSERTION OF SHUITLE PLASMID DNA INTO THE HSV-1 GENOME 5511 waspurified byrepeated roundsoflimiting dilution and dot
blot analysis. The laxP insertion into the tk locus was
confirmedbySouthernblotanalysis(see Fig. 2) (30).Nytran filterscontainingtheelectrophoretically separated EcoRI or BamHIfragmentfromthe indicated DNAs were firstprobed withatk probe(2,416-bpEcoRI fragmentfrom pUCXI)(see
Fig. 3), strippedto remove the tk probe, and rehybridized
with aloxPprobe(350-bpNaeI-SmaI fragment frompBS64) (see Fig. 3). All probes used for dot blot and Southern analyseswerelabeledwith a Random Primed DNALabeling Kit(Boehringer Mannheim Biochemicals).
InvitroCre-mediated recombination. loxPshuttleplasmids
wererecombinedinto the KAT::loxlgenome in cell-freeCre reactions under conditions similar to those described by
Abremski andHoess(1)asmodifiedbySauer et al. (28). The DNAmixturewasincubatedat30°C for 30 to 40mininthe
presence of Cre buffer (50 mMTris-HCl
[pH
7.5], 33 mMNaCl,10mMMgCl2)and 40 ngofpurified Creprotein (NEN Dupont Inc.,Wilmington, Del.). Forintermolecular recom-bination, reaction mixtures included KAT::laxl viral DNA
and a 20- to 50-fold excess of shuttle plasmid DNA, and
polyvinyl
alcoholwasaddedto afinal concentration of 1.7%toenhanceintermolecular recombination (28,35). Recombi-nationreactionswereterminatedbyheatingsamples to 65°C
for 10 min, and reactionmixtures weretransfected into D6
cells to produce infectious virus. When recovery of the
shuttle plasmid from viral DNA was required, purified
KAT::108 viral DNA wasincubatedwithCrein theabsence ofpolyvinyl alcohol andtherecovered plasmidwas used to
transform DH5a by standardprotocols (27).
PurificationofCre-mediated recombinantviruses. Recom-binant viruses were identified on the basis of their
blue-plaque phenotype and purified by plaque purification and
limiting dilution. Transfection lysates were diluted and
platedatlowmultiplicity of infection (<0.01)ontoconfluent
D6 cell monolayers. Plaques weretested for lacZ activity with Bluo-gal
(GIBCO-BRL),
and viruses producing blue plaqueswerepickedas anagaroseplugand resuspended inphosphate-buffered
saline (PBS). Individual plaque isolateswerethencloned bylimiting dilutionon D6cellsin
microti-terplates.Viruses wereconsidered pure when100% of the
plaques stained blue through three successive rounds of limiting dilution.
Assays for lacZ
activity.
Two assays wereused to detect lacZ activity indeveloped
plaques.
For isolation ofviable virus, cultureswere rinsed withPBS(10
mMsodiumphos-phate
[pH
7.1], 136 mMNaCl),
overlaid with 300 mg ofBluo-gal-0.5% low-melting-point
agarose(GIBCO-BRL)
perml in MEM
supplemented
with10mM HEPES and5%fetal calf serum, and incubated at37°C
until colordeveloped.
When isolation of viable virus was not
required,
infected cultureswerefixed andstainedby
ahistologicalmethod with5-bromo-4-chloro-3-indolyl-,3-D-galactopyranoside
(X-Gal)
(Boehringer
MannheimBiochemicals).
Infected cultures wererinsedwithPBS,
fixedfor 5 minatroomtemperaturein2%
formaldehyde-0.3% glutaraldehyde
inPBS,
andincu-batedin a
chromophore
solutioncontaining
0.1%X-Gal,
5mM
K4Fe(CN)6
3H20,
5mMK3Fe(CN)6,
and 2 mMMgCl2
in PBS. Incubation continuedat
37°C
until colordeveloped
(usually after30to60
min),
and themonolayerswereagain
rinsedwith PBS.
RESULTS
Direct isolation of
Crelox-derived
recombinant viruses onthebasis of
13-gal
reporter activity. Thegeneral
strategyforK&T:oxi 03i -0
h.P,
1K
kb
-~~~~3~' 4.
bRa M13
wt
acMv-IaeZ~~~CI
1L:
1amP at gB HCMV-iac2 o*rPba MI3 kW
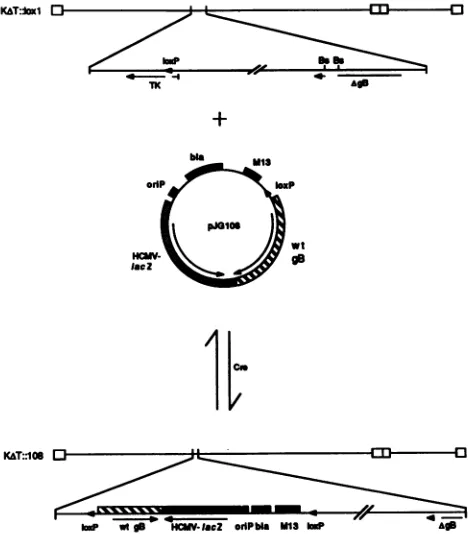
FIG. 1. HSV-basedCre-loxsite-specific recombination system. The target virus for site-specific recombination,
KAT::lox1,
was derived from KAT, and its genome contained (i) acis-acting site-specific recombination site laxP from bacteriophageP1(arrowhead) inserted into the tk coding sequence and (ii) a defective gB allele (AgB) due to a 969-bpBstEII
(Bs) deletion in the gB coding sequence, including the transmembrane domain. In the example shown, thepJG108 shuttle plasmid was recombined intoKAT::laxl.
pJG108containedalaxP recombination site(arrow),an HCMVIEP-lacZ reporter gene cassette for identification of virus recombinants, and thewild-type gB allele from HSV-1 strain KOS. The polarities of both the viral and theplasmidlaxP sites are shown. pJG108 was inserted into the
KAT::loxl
genomeatthe tklaxPsite inacell-free Cre-mediated recombination reaction, and infectious virus was produced by transfection of the reaction mixture into D6 cells (8). The recombinant KAT::108 virus was diploid for the gB gene, containing both the deletedKAT::loxlallele(AgB)
andthe wild-type KOS allele (wt gB). KAT::108 also yielded blue plaques in the presence of Bluo-gal or X-Gal because of the HCMVIEP-lacZ reporter gene.using an HSV-1 shuttlevector systembased on the
bacte-riophage
P1
Cre-lox
site-specific
recombinationmachinery
is shown in Fig. 1. The system consists of arecipient
virus genome and ashuttleplasmid,
eachcontaining
afunctional cis-acting laxPrecombinationsite,
andtheCrerecombinaseprotein.
In acell-freereaction,
Cre-mediated
recombinationoccurs
directly
between the twoloxP sites(1)
such that the entireplasmid
is inserted into therecipient
viral genome.Because the Crereaction is
reversible,
the shuttleplasmid
can also berecovered from the viralgenome.
A recipient target
virus,
KAT::loxl,
was derivedby
markertransferofafunctionalloxP site into the tk locus of the gB- HSV-1 mutant KAT genome
(8).
LikeKAT,
theKAT::loxl
recipientviruscontainsa969-bp
BstEII deletionwithintheessentialgB gene andmustbe
propagated
onthegB-complementing
D6cell line(8).
KAT::loxlgenomic
DNA wasanalyzed
bySouthern
blot(30)
toconfirmthepresenceof the
loxP
site within the tkcoding
sequence(Fig. 2).
Shuttle
plasmids
containing
a functional loxPsite,
are
- F--VOL.66,1992
KATT108rl
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.326.560.75.342.2]J. VIROL.
bla bla
M13 oriP M13 oriP Kpnl
BamHl
: loxP BamHI
(BcllBamHt)
pJG108 pJG102
11.69kb 12.86kb
HCMV- 9B-1 HCMV- gB-1
_acZ (KOS321) /acZ (tsBS)
[image:4.612.64.295.78.432.2]BaBamHamH lo Kpnl
FIG. 3. pJG108and pJG102 shuttle plasmids. Each plasmidwas derived frompBS+ and containsaloxP site-specific recombination site,anHCMVIEP-lacZreportergene,andanHSV-1gBgeneallele as thetestgene.ThegBgenes ofpJG108andpJG102arederived fromHSV-1 strains KOS andtsB5, respectively. Restriction sites usedtoclonetherespective fragmentsareindicated.
:- ..) to - a go
C
.1 .-: 'jr] E
K \\T
_P-.. 4;:
FIG. 2. Southernanalysisof KAT::loxlviral DNA. Pla
viral DNAs were digested as indicated, electrophorese
agarose, and transferred to a Nytran filter(Schleicher &
Keene, N.H.). The filter was sequentially hybridized i
labeledprobestotk(2,416-bpEcoRIfragmentfrompUCXI
the 1oxP-containing fragment (364-bp NaeI-SmaI fragm pBS64) (B). The tk probe was quantitatively removed hybridizationwith the loxPprobe. (C)Restrictionmapsof regions. Enzymes used were BamHI (B) and EcoRI positionsof the tkgene (arrow)and the tk SnaBI(Sn)sit
clone the 354-bp SmaI (S)-NaeI (N) frompBS64are als
Expected restrictionfragmentsizes in kilobasesarealsos
porter gene consisting of the bacterial lacZgene u:
transcriptional control of the strong HCMVIEP-e element, and one oftwo different alleles of the H'
genewereconstructed frompBSM13- (Fig. 3). The
gene was included for use as a marker for idt
recombinant virusescontaining the inserted shuttle I pJG108 contained the complete gB genefrom theM
HSV-1 strainKOS. pJG102 contained the HSV-1str mutantgBgene;themutantproteinproduct has bee tobedefectiveat 39°C andto induce polykaryocyt tion, or the syncytial (syn) phenotype, in infected
34°C (20).
The pJG102 and pJG108 shuttle plasmids were i
dently introduced into the KAT::Ioxl genome ina
Cre reaction. Infectious viruswasproduced bytran oftheCre reaction mixtures into the gB-complemei cell line. After 2 days, transfection lysateswere h.
andreplated on D6 cells andplaqueswere tested for P-gal
activity. Virus from individual blue plaques from each
ex-perimentwasisolated and subsequently clonedon D6cells by multiple roundsof limiting dilutions. Viruseswere
con-sidered pure when 100% of the isolates testedwere P-gal+
through three rounds of limiting dilutions. For each virus, '1''' insertion of the shuttle plasmidwasconfirmed bySouthern blot analysis (data not shown). Recombinant viruses were
namedby combiningthe KAT::prefixof the recipientvirus with the numeric suffix of the shuttle plasmid to yield KAT::102 andKAT::108, respectively.
__-__I Cre-mediated recombinationishighlyefficient. One
appeal-ing
property
of theCre-lox
system
forintroducing
DNA into the pseudorabies virus genome was the relatively high recombination efficiency achieved (28). Itwas therefore of l_ interest to determine the recombination efficiency of the HSV-basedsystem. Thepresenceof the lacZreportergeneon the inserted shuttle plasmid provided a convenient
smid and marker for
identifying
recombinant virusesagainst
aback-d in 1% ground of nonrecombinant viruses. SinceCrereaction
mix-Schuell, turesweretransfected into the
gB-complementing
D6cells,
with 32p_ both the nonrecombinant gB- KAT::loxl and the
Cre-1)(A) and mediated gB+ recombinants should undergo productive
rep-ent from lication with essentially equal efficiencies. Therefore, the prior to percentage of P-gal+ plaques produced on D6 cells from a frelevant given transfectionlysate provided areliable estimate of the
(E). The recombinationefficiency ofaparticular recombination
reac-eused to tion mixture.
shown.
DNA from fourindependent preparationsofpJG108
was used in separate Cre reactions witha single preparation ofKAT::loxl viral DNA, and viruses in the resulting transfec-tion lysates were examined for frequencies of recombina-nderthe tion. The resultsarereportedasthe number of blue plaques
SV
gB-n
per total number of plaques (Table 1). The Cre-mediated reporterentifying
plasmid. TABLE 1. Cre-mediated recombination efficiency vild-type
rain tsB5
nshown e forma-lcells at indepen-cell-free isfection
nting D6 arvested
Plasmid' Dilution' No.of blue No. oftotal % blue
plaques plaques
plaquesc
None -7 0 120 0
pJG108 -5 8 40 20
pJG108 -5 5 36 14
pJG108 -4 26 233 11
pJG108 -5 5 41 12
a Cre reactions containedeitherno shuttle plasmid DNA or DNA from one
of fourpJG108 plasmidDNA stocks.
bDilution oftransfected cell lysate used for blue-plaque assay.
cPercentblue-plaquevalues are (blue plaques/total plaques) x 100.
5512 GAGE ET AL.
A
B
K,tS cl1
t !.,.
!'.., lp isa iza.-a
41',.f)p 0
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.318.555.78.171.2] [image:4.612.318.555.630.703.2]INSERTION OF SHUTTLE PLASMID DNA INTO THE HSV-1 GENOME TABLE 2. Rescue of gB- phenotype ofKAT::1oxl by
Cre-mediated insertion ofpJG108 orpJG102
Titer(PFU/ml) D/eo Plaque
Virus Titer_(PFIJ/ml)_ D6/Vero morphology
efficiency
D6 Vero D6 Vero
KAT::laxl 1.8 x 108 0 0 Syn+ NAb
KAT::108 8.5 x 108 9.1 x 108 1.07 Syn+ Syn+ KAT::102 1.7 x 107 1.8 x 107 1.06 Syn Syn
a For each virus, efficiencyis defined as thetiteron Verocells/thetiter on
D6 cells.
bNA,notapplicable.
recombination efficiencies ranged from 11 to 20% and aver-aged about 15%.
Complementation of the gB-
phenotype
of KAT::Ioxl. Experimentswere carried out to confirm that expression ofthepolypeptidefromthe shuttle plasmid would complement the defective gB gene at its nativeviral locus and that the
phenotype ofthe resulting recombinant virus would reflect thegenotype of the plasmid copy of the gene in thetklocus.
KAT::108and KAT::102 both encode functional gB
polypep-tides on their respective shuttle plasmids, with KAT::108
encodingthewild-type gB polypeptide and KAT::102
encod-ing themutant gB polypeptide from HSV-1 strain tsB5. The
tsB5 gB polypeptide includes a ts mutation within the
external domain (10) and a syn mutation (Arg to His) at
residue 858within the cytoplasmic domain (5, 6, 10).
There-fore, KAT::108 andKAT::102 were used to demonstrate (i)
that complementation of the gB- defect of the parental
KAT::loxl virus after insertion of the pJG108 or pJG102 shuttle plasmid occurred and (ii) that the tsB5 mutant gB
polypeptide encoded by pJG102 conferred both mutant
phenotypes (temperature sensitivity and syncytial plaque
morphology)onKAT::102.
In an initial experiment, both the KAT::108 and the
KAT: :102 viruseswereshowntoreplicate normallyon Vero
cells inplaqueassays (datanotshown). Therefore, titersof
KAT::108 and KAT::102 stocks preparedonVerocells anda
KAT:
:loxl
stockgrown on D6cellsweredeterminedonbothD6andVerocells.Asexpected,
KAT::lQxl
developedatiteronthegB-complementingD6cell line butnot onVerocells (Table 2). In contrast, KAT::108 and KAT::102 each grew
equally well on either cell line.The plaque
morphology
of each virus was also examined. KAT::102produced
the mutantsynplaquemorphology
onboth cell lines(Table 2).
Therefore,
expression
offunctional gBpolypeptides
from the inserted shuttleplasmids complemented
the deletion within thegBgeneatits native virallocus andexpression
ofthe allele from
pJG102
conferred the synphenotype
on KAT::102. The KAT::102recombinant viruses alsodemon-strated temperature-dependent virus
growth.
The ratio of KAT::102 recombinant virusproduced
at 39°Ccompared
with that
produced
at34°Cwasapproximately
5 x10-4,
an indexof temperaturesensitivity
similartothat of theparent virustsB5.KAT::108grewequallywellatboth 34 and39GC.
Efficient Cre-mediated recovery of intact shuttle
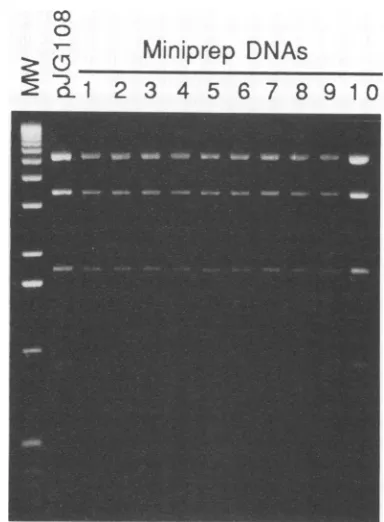
plasmids
from KAT::108 viral DNA. Sauer et al.
(28)
demonstrated thatinserted shuttleplasmid
DNAcouldbeexcised fromthepseudorabiesvirus genome in a
Cre-dependent
mannerbutdid notexamine the
integrity
of the recovered shuttleplas-mid. If the
Cre-lax
methodology
is to be used as a true shuttlevector system, the inserted shuttleplasmids
recov-ered from the viral genome must be intact. The shuttle
a0
Miniprep DNAs
2
3
4
5
6 7 8
9 10
FIG. 4. Cre-mediated rescue of intact shuttle plasmid DNA from KAT::108 viral DNA. KAT::108 viral DNA was reacted with Cre recombinase and transformed intoDH5a to propagate the rescued shuttleplasmid. Miniprep DNAs were isolated from 10 independent colonies, restricted withPstI, andelectrophoresed in 1% agarose. All10miniprep DNAs are identical to the pJG108 control.
plasmid was therefore recovered from extracted KAT::108 viralDNA and compared with the original pJG108plasmid stock by restriction enzyme analysis. The plasmid was
released frompurified KAT::108DNAin anintramolecular Crereaction andpropagated after transformation ofDH5a. Small batches of
plasmid
DNA(17) were prepared from 10isolated colonies, and their PstI restriction patterns were
compared with that of pJG108. All 10 miniprep DNAs
showed restrictionfragment
patterns
that were identical tothat of
pJG108
(Fig. 4), demonstrating
that large shuttleplasmids couldbe recovered intact from the viral genomeby Cre-mediated recombination.
DISCUSSION
In this report, we describe the
adaptation
of the Cre-lax site-specific recombination system of bacteriophage P1 for usewith HSV-1 andinvestigate
thepotential
usefulness ofthis
approach
for thegenetic
analysis
of an essential viralgene.
Site-specific
recombination mediatedby
the Crere-combinase
protein
occurredbetweentwoloxPsites,
oneon the shuttleplasmid
and the other on therecipient
virus genome. Aconstitutively
expressed
lacZ reporter gene cassette on the shuttleplasmids
allowedeasy identification of recombinant viruses on the basis of theirblue-plaque
phenotype. Because Cre-mediated recombination was
re-versible,the
plasmids containing
clonedcopies
of the essen-tialgBgene could berecoveredfor furtheranalysis by
using
a second Cre reaction. Recovered shuttle
plasmids
had arestriction fragment
pattern
identical to that of theoriginal
plasmid.
Expression
offunctionalgB
polypeptides
from the inserted shuttleplasmids
at the tk locuscomplemented
aVOL.66, 1992 5513
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.60.305.94.176.2]5514 GAGE ET AL.
lethal defect in thegBgene. Expression of the tsB5gBallele conferred the ts and syn phenotypes on the recombinant
virus,
demonstratingthat thisapproach is useful fortesting
mutantconstructs foraltered phenotypes.
TheCre-loxshuttlesystem offers anumber of
advantages
fordetailed
genetic
analysescompared with a strategy based on markertransfer. (i) Since recombination occursthrough
the
34-bp
cis-acting loxP site, no homologous viralflanking
sequencesarerequiredtofacilitaterecombination,
minimiz-ing clonminimiz-ing
steps that may be needed for marker transfer. Moreover, this site-specificrecombinant event avoidspoten-tial
problems
related to rearrangement of theintegrated
DNA which sometimes occurs in marker transfer
experi-ments.This isparticularlyimportant in experiments
involv-ing
the insertion of large DNA fragments. Although theCre-lox approach
does introduce plasmid DNA sequencesalong
with the testgene(s),thefunction of either the reporter or the gB gene was not affected by the presence of these nonviral sequences. (ii) Therecombination frequenciesob-served for the HSVCre-lox system were even higher than those
reported
forthepseudorabiesvirus-based system and areasmuchas 1 order ofmagnitude (10to 20% versus<2%)greater than those typically observed in marker transfer
experiments.
The constructs containedboth a complete test gene and a reporter gene such that recombinants can bereadily
detected andpurified
on the basis of reporter geneexpression.
Also,inthese studies, theexpectedcomplemen-tation of the deleted gB allele by the plasmid-encoded
wild-type
and tsB5 gB proteins could have been used as a means ofselecting for recombinants by transfection oftheCre-mediated
recombination reaction mixtures directly intononcomplementing
Vero cells. (iii) Unlike DNA introducedby
markertransfer,genesintroducedonshuttle plasmids aredirectly
recoverable from theviralgenomein a cell-free Cre reaction without additional cloningsteps and are thus readilyavailable for further analysis. Thisis particularly important for studies requiring the generation and testing of a large
panel
of mutants. For example, randomly mutagenized genescanbeintroduced intotheviral genome and tested for aparticular
phenotypesuch assyncytialplaque morphology, and then only those mutant constructs demonstrating thedesired
phenotype
may be recovered from plaque-purified viral DNA for analysis by DNA sequencing. Thus, theability
to easily recover inserted shuttle plasmids from the viralgenomeshouldmake a priordetailed analysisof genetic constructs unnecessary. (iv) Finally, theCre-lox-based
re-combination system should be useful for viral systems for
which marker transfer procedures are currently difficultor
unavailable. These includethecytomegaloviruses, Epstein-Barrvirus, andvaricella-zoster virus.
ACKNOWLEDGMENTS
Wethank Neal DeLuca,William Goins, RameshRamakrishnan, and Stan Person forsuggestions,discussions of the data, andcritical readingof themanuscript.
Thiswork was supportedby U.S. Public Health Service grants GM34534, A126937, and A118228 from the National Institutes of Health.
REFERENCES
1. Abremski, K., and R. Hoess. 1984. Bacteriophage P1 site-specific recombination: purification and properties ofthe Cre recombinase.J. Biol. Chem. 259:1509-1514.
2. Abremski,K., R.Hoess, and N.Sternberg.1983. Studies on the properties ofP1site-specific recombination:evidence for topo-logically unlinked products following recombination. Cell 32: 1301-1311.
3. Arsenakis, M.,L.FoaTomasi,V.Speziali, B.Roizman,andG. Campadelli-Fiume. 1986.Expressionandregulationof
glycopro-tein C geneofherpessimplexvirus1residentin aclonal L-cell line. J.Virol. 58:367-376.
4. Blin, N., and D. W. Stafford. 1976. A general method for isolation of high molecular weight DNA from eukaryotes.
Nucleic Acids Res. 3:2303-2308.
5. Bzik, D. J., B. A. Fox, N. A. DeLuca, and S. Person. 1984. Nucleotide sequence of a region of the herpes simplexvirus type 1 gB glycoprotein gene: mutations affecting rate of virus entry and cell fusion. Virology137:185-190.
6. Cai, W.,B.Gu,andS. Person. 1988. Roleofglycoprotein Bof herpes simplexvirus type 1 in viral entry and cell fusion. J. Virol. 62:2596-2604.
7. Cai, W., S. Person, C. Debroy, and B. Gu. 1988. Functional regions and structural features of thegBglycoprotein ofherpes simplexvirus type1:an analysisoflinker insertionmutants. J. Mol. Biol. 201:575-588.
8. Cai, W., S.Person, S. C. Warner, J. Zhou,andN. A. DeLuca. 1987. Linker-insertion nonsense and restriction-site deletion mutations of the gBglycoprotein gene of herpes simplex virus type 1. J.Virol. 61:714-721.
9. Costa,R.H.,K.G.Draper, G. Devi-Rao,R. L.Thompson, and E. K. Wagner. 1985. Virus-induced modification of the host is required for expression of the bacterial chloramphenicol acetyl-transferase gene controlled by a late herpes simplex virus promoter(VP5). J. Virol. 56:19-30.
10. DeLuca,N.A.,D.J. Bzik,V.C.Bond, S. Person, andW.Snipes. 1982. Nucleotide sequences of herpes simplex virus type 1 affectingvirus entry,cell fusion, and production ofglycoprotein gB (VP7). Virology122:411-423.
11. DeLuca, N. A., A. M. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558-570.
12. Dennis, D., and J. R. Smiley. 1984. Transactivation of a late herpes simplex virus promoter. Mol. Cell. Biol. 4:544-551. 13. Graham,F.L.,andA. J.vander Eb.1973.Anew techniquefor
the assay of infectivity of human adenovirus5DNA. Virology 52:456-467.
14. Gross-Bellard, M., P. Oudet, and P. Chambon. 1973. Isolation of high molecular weight DNA from mammalian cells. Eur. J. Biochem. 36:32-38.
15. Hanahan, D. 1983. Studies on transformation ofEscherichia coli withplasmids. J. Mol. Biol. 166:557-580.
16. Hoess, R., and K. Abremski. 1985. Mechanism of strand cleav-age and exchange in the Cre-lox site-specific recombination system. J. Mol. Biol. 181:351-362.
17. Holmes, D. S., and M. Quigley. 1981. A rapid boiling method for the preparation of bacterial plasmids. Anal. Biochem. 114:193-197.
18. Homa, F. L., T. M. Otal, J. C. Glorioso, and M. Levine. 1986. Transcriptional control signals of a herpes virus late
(Y2)
gene lie within bases -34 to +124 relative to the 5' terminus of the mRNA.Mol. Cell. Biol. 6:3652-3666.19. Krikos, A. Unpublished results.
20. Manservigi, R., P. G. Spear, and A. Buchan. 1977. Cell fusion inducedbyherpessimplex virus is prompted and suppressed by differentviralglycoproteins. Proc. Natl. Acad. Sci. USA 74: 3913-3917.
21. McGeoch, D. J., M. A. Dalrymple,A. J. Davidson, A. Dolan, M. C. Frame, D. M. McNab, L.
Perry,
J. E. Scott, and P. Taylor. 1988.ThecompleteDNA sequence of the long unique region of the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531-1574.22. McGeoch, D. J., A. Dolan, S. Donald, and D. H. K. Brauer. 1986. Complete DNA sequence of the short repeat region of herpes simplex virus type 1. Nucleic Acids Res. 14:1727-1745. 23. McGeoch, D. J., A. Dolan, S. Donald, and F. J. Rixon. 1985. Sequence determination and genetic content of the short unique region in the genome of herpes simplex virus type 1. J. Mol. Biol. 181:1-13.
24. Post, L. E., and B. Roizman. 1981. A generalized technique for J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
INSERTION OF SHUTTLE PLASMID DNA INTO THE HSV-1 GENOME 5515 deletion ofspecificgenesinlargegenomes:agene22 ofherpes
simplexvirus 1 isnotessentialfor growth. Cell 25:227-232. 25. Roizman, B. 1982. The family herpesviridae: general
descrip-tion,taxonomy,and classification,p.1-23. In B. Roizman (ed.),
Theherpesviruses 1. Plenum Press, New York.
26. Roizman, B., and F. J. Jenkins. 1985. Genetic engineering of novelgenomesof largeDNAviruses. Science 229:1208-1214.
27. Sambrook, T., E. F. Fritsch, and T. Maniatis. 1982. Molecular cloning:alaboratory manual,2nd ed. ColdSpringHarborPress,
ColdSpring Harbor,N.Y.
28. Sauer, B., M. Whealy, A. Robbins, and L. Enquist. 1987. Site-specificinsertion of DNA intoapseudorabiesvirusvector. Proc. Natl. Acad. Sci. USA 84:9108-9112.
29. Silver, S.,and B.Roizman. 1985. y2-thymidinekinase chimeras
areidenticallytranscribed butregulatedas-y2genesin herpes simplexvirusgenomes and as genesin cell genomes. Mol. Cell. Biol. 5:518-528.
30. Southern, E. M. 1975. Detection ofspecific sequences among DNAfragments separated by gel electrophoresis. J.Mol. Biol.
98:503-517.
31. Southern,P. J., andP. Berg.1982. Transformation of
mamma-lian cellstoantibiotic resistancewithabacterialgeneunder the
controlof theSV40 early regionpromoter.J.Mol. Appl. Genet. 1:327-341.
32. Spaete, R. R., and E.S. Mocarski. 1985. Regulation of
cytomeg-alovirusgeneexpression:aand promotersaretransactivated by viral functions in permissive human fibroblasts. J. Virol. 56:135-143.
33. Sternberg, N., and D. Hamilton. 1981. Bacteriophage P1 site-specificrecombination. 1. Recombination between loxPsites. J. Mol. Biol. 150:467-486.
34. Stinski, M. F., and T.J.Roehr. 1985. Activation of themajor immediate earlygene of humancytomegalovirus by cis-acting
elements in the promoter-regulatory sequence and by
virus-specific trans-actingcomponents.J. Virol. 55:431-441. 35. Tanford, C. 1961. Physical biochemistry of macromolecules.
Wiley,New York. VOL. 66, 1992