Copyright © 2002, American Society for Microbiology. All Rights Reserved.
Combination of Drugs and Drug-Resistant Reverse Transcriptase
Results in a Multiplicative Increase of Human Immunodeficiency
Virus Type 1 Mutant Frequencies
Louis M. Mansky,
1,2,3* Dennis K. Pearl,
3,4and Lisa C. Gajary
2,3Department of Molecular Virology, Immunology, and Medical Genetics,1Department of Statistics,4Center for
Retrovirus Research,2and Comprehensive Cancer Center,3Ohio State University Medical Center,
Columbus, Ohio 43210
Received 31 January 2002/Accepted 15 June 2002
Replication of drug-resistant human immunodeficiency virus type 1 (HIV-1) in the presence of drug can lead to the failure of antiretroviral drug treatment. Drug failure is associated with the accumulation of drug
resistance mutations. Previous studies have shown that 3ⴕ-azido-3ⴕ-deoxythymidine (AZT), (ⴚ)2ⴕ,3ⴕ
-dideoxy-3ⴕ-thiacytidine (3TC), and AZT-resistant HIV-1 reverse transcriptase (RT) can increase the virus in vivo
mutation rate. In this study, the combined effects of drug-resistant RT and antiretroviral drugs on the HIV-1 mutant frequency were determined. In most cases, a multiplicative effect was observed with AZT-resistant or AZT/3TC dually resistant RT and several drugs (i.e., AZT, 3TC, hydroxyurea, and thymidine) and led to increases in the odds of recovering virus mutants to over 20 times that of the HIV-1 mutant frequency in the absence of drug or drug-resistance mutations. This observation indicates that HIV-1 can mutate at a signif-icantly higher rate when drug-resistant virus replicates in the presence of drug. These increased mutant frequencies could have important implications for HIV-1 population dynamics and drug therapy regimens.
The treatment of human immunodeficiency virus type 1 (HIV-1)-infected individuals with antiretroviral drugs includ-ing reverse transcriptase (RT) and protease inhibitors in a combination therapy (called highly active antiretroviral ther-apy, or HAART) has significantly reduced the rate of HIV-and AIDS-related morbidity HIV-and mortality (34, 37). However, a problem with these therapies is that they can be suboptimal, due in some cases to a lack of patient compliance to drug administration (2, 12). Suboptimal drug treatment can lead to the selection of drug-resistant viruses which can limit the clin-ical benefit of drug treatment and even lead to new variant viruses with altered virulence and tropism (4, 18, 32). Clinical drug resistance to RT inhibitors such as 3⬘-azido-3⬘ -deoxythy-midine (AZT) and (⫺)2⬘,3⬘-dideoxy-3⬘-thiacytidine (3TC) is commonly conferred by single (3TC) or several (AZT) amino acid changes in RT.
The in vivo mutation rate for HIV-1 was previously deter-mined to be 4 ⫻ 10⫺5 mutations per target base pair per
replication cycle (22, 28), which predicts that about one muta-tion occurs for every three new genomes produced. Thus, viral genomes with each possible mutation as well as many with double mutations are likely generated each day. When drug treatment incompletely suppresses viral replication, the selec-tion and fixaselec-tion of mutaselec-tions that confer drug resistance oc-curs at a rapid rate (38, 40). These drug-resistant viruses can readily reside in latently infected cells, which further compli-cates subsequent drug treatment regimens during the life of the infected individual (6, 42). When drug resistance mutations accumulate, drug susceptibility diminishes and reduces the
po-tency of the components of HAART. Continued replication in the presence of drug will select for even greater levels of resistance and typically leads to cross-resistance to drugs of the same class (17, 39). Transmission of HIV-1 with reduced sus-ceptibility to antiretroviral drugs may compromise the efficacy of drug therapy (10).
Several previous studies have investigated how antiretroviral drugs can influence the fidelity of retrovirus replication. For instance, 5-azacytidine, which is a nucleoside analog that is incorporated into RNA and inhibits protein synthesis, was previously found to increase the in vivo mutation rate of spleen necrosis virus (SNV; an avian retrovirus) by a factor of 13 (33). AZT was subsequently observed to increase the SNV mutant frequency by a factor of 10, and AZT was found to increase the mutant frequency of murine leukemia virus (MLV) by a factor of 3 (15). Another study showed that deoxynucleoside triphos-phate (dNTP) pool imbalances created by treating cells with either hydroxyurea (HU) or thymidine (Thy) can significantly increase the SNV and MLV mutation rates but that the influ-ence of AZT on the SNV and MLV rates did not involve altering dNTP pools (16).
A recent study investigated how AZT and 3TC, as well as AZT and 3TC resistance-conferring mutations, influence the in vivo mutation rate of HIV-1 (25). This analysis utilized the lacZ␣peptide gene as a mutational target, as in other studies (23, 25–28). AZT increased the HIV-1 mutation rate 7.6-fold in a single round of replication. In addition, 3TC increased the virus mutation rate 3.4-fold. AZT-resistant RT was also found to influence the mutation rate. In particular, replication of HIV-1 with AZT-resistant RTs increased the mutation rate as much as 4.3-fold, while replication of HIV-1 with a 3TC-resis-tant RT had no significant effect on the mutation rate. It was observed that only high-level AZT-resistant RT variants could * Corresponding author. Mailing address: Department of Molecular
Virology, Immunology, and Medical Genetics, Ohio State University, 2078 Graves Hall, 333 W. 10th Ave., Columbus, OH 43210. Phone: (614) 292-5525. Fax: (614) 292-9805. E-mail: [email protected].
9253
on November 8, 2019 by guest
http://jvi.asm.org/
influence the in vivo mutation rate (i.e., those containing the mutations M41L/T215Y and M41L/D67N/K70R/T215Y).
In this study, the combined effects of drug and drug-resistant virus were analyzed. It was found that the replication of AZT-resistant HIV-1 in the presence of AZT led to a multiplicative increase in the odds of recovering mutants to over 20 times that observed with wild-type (wt) virus in the absence of drug. Furthermore, replication of AZT/3TC dually resistant virus in the presence of AZT and 3TC also led to a multiplicative increase in the odds of recovering virus mutants. Finally, the replication of AZT-resistant virus in the presence of HU and Thy also increased virus mutant frequencies.
MATERIALS AND METHODS
Retroviral vectors and expression plasmids.The HIV shuttle vector used in these studies has been previously described (23, 27, 28). The vector cassette contains the SV40 promoter driving expression of the neomycin
phosphotrans-ferase gene (neo), a bacterial origin of replication from pACYC 184, and the
lacZ␣peptide gene (Fig. 1A). The HIV-1gag-polexpression plasmid, the
am-photropic murine leukemia virusenvexpression plasmid, and the vector used for
expression of wt Vpr have been previously described (27). The RT variants analyzed in these experiments were constructed by introducing mutations en-coding RT amino acid substitutions into pSVgagpol-rre-r by a primary and combinatorial two-step PCR protocol (14, 23).
Transfections, infections, and cocultivations.The COS-1 and HeLa cell lines used were obtained from the American Type Culture Collection (Rockville, Md.) and were maintained in Dulbecco’s modified Eagle’s medium containing 10% calf serum or 10% fetal bovine serum, respectively. HIV-1 vectors and expression plasmids were transfected into HeLa cells by use of Superfect (Qiagen). HeLa cells were infected in the presence of Polybrene (13). Infection of HeLa target cells was also done by cocultivation of virus-producing cells with target cells (24, 29).
The influence of the antiretroviral drugs on HIV-1 mutant frequencies was determined by posttreatment of cells with drug. Posttreatment refers to
main-taining HeLa target cells in medium supplemented with drug for 2 h before cocultivation and continued until 24 h after cocultivation. Posttreatment with drug influences the HIV-1 mutant frequency only during reverse transcription (25).
Experimental protocol for single cycle of HIV-1 replication.The experimental protocol developed to obtain a single cycle of HIV-1 shuttle vector replication is shown in Fig. 1 and described in detail elsewhere (23, 27, 28). Proviral DNA containing the mutation target was purified with the Lac repressor protein as
previously described (29). The Lac repressor protein was purified from
Esche-richia colistrain HB101/lacpIQ (kindly supplied by Tom Record, University of Wisconsin, Madison) as previously described (19). The purified proviral DNA
was introduced intoE. coli, and the ratio of white plus light-blue bacterial
colonies to total bacterial colonies observed provided a forward mutant fre-quency for a single HIV-1 replication cycle.
RESULTS
One round of HIV-1 vector replication.The general protocol
developed to assay a single round of HIV-1 shuttle vector replication is shown in Fig. 1. Cocultivation was used to pro-duce infected target cells in order to obtain the largest number of infected cells for analysis of the mutant frequency. Coculti-vation of mitomycin C-treated step 2 cells (typically 2.5⫻105
cells per 60-mm-diameter petri dish, 5⫻105cells per
100-mm-diameter petri dish, or 7.5⫻ 105 cells per 150-mm-diameter
petri dish) with fresh HeLa target cells led to 8⫻102to 3⫻
103CFU/2.5⫻105HeLa target cells in the absence of drug.
The steps going from a parental shuttle vector provirus in the virus-producing cell to a vector provirus in the target cell con-stitute a single cycle of replication (Fig. 1). The relative amount of infectious virus produced from the virus-producing cells was assessed by monitoring the number of G418-resistant colonies obtained from cocultivations.
FIG. 1. HIV-1 vector used for analysis of virus mutant frequencies. (A) Expression cassette. The HIV-1 vector used has been previously described (23, 27, 28). The vector contains a cassette with the simian virus 40 promoter driving expression of theneogene, a bacterial origin of replication, and thelacZ␣peptide gene. (B) Protocol for one cycle of HIV-1 vector virus replication. The steps going from a parental shuttle vector provirus in the step 2 cell to a vector provirus in the step 3 cell constitute a single cycle of replication. The influence of the antiretroviral drugs on HIV-1 mutant frequencies was determined by posttreatment of step 3 cells with drug. Posttreatment refers to maintaining step 3 cells in medium supplemented with drug for 2 h before cocultivation and continuing until 24 h after cocultivation. G418-resistant cells infected with the HIV-1 vector were pooled and the total DNA was purified and digested with restriction enzymes. The vector cassette was selectively purified with the Lac repressor protein, ligated, and introduced intoE. coli. The ratio of light blue and white bacterial colonies to total colonies observed was used to determine virus mutant frequencies.
on November 8, 2019 by guest
http://jvi.asm.org/
The influence of drugs together with drug-resistant HIV-1 RT on the rate of HIV-1 mutation was determined by main-taining the HeLa target cells in medium supplemented with drug. The target cells were treated for 2 h before infection as well as 24 h after infection. The drug treatments were initiated 2 h before infection to ensure that dNTP pools were altered by the drug at the time of infection, when the process of reverse transcription initiates. The target cells were maintained in me-dium with drug for 24 h after infection to ensure that the dNTP pool imbalance was present throughout HIV-1 replication and establishment as a provirus (⬃8 h after infection). The drug concentrations used were chosen based on physiological rele-vance, previous observations, and concentrations that would not greatly diminish the level of infection of target cells (15, 16, 25, 31).
AZT or 3TC and AZT-resistant HIV-1 RT together result in a multiplicative increase in the odds of recovering virus
mu-tants. AZT and AZT-resistant HIV-1 RT have individually
been shown to increase mutant frequencies during HIV-1 rep-lication (25). Since during the course of antiviral therapy there are HIV-1-infected individuals harboring viruses with drug re-sistance mutations, the combined effect of drugs and drug resistance mutations on the HIV-1 mutation rate is of interest. The analysis of the replication of AZT-resistant HIV-1 in
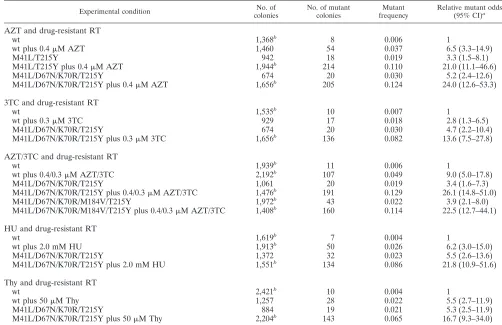
[image:3.587.39.541.84.409.2]the presence of AZT could lead to several possible effects on the mutation rate, including additive, multiplicative, synergis-tic, and antagonistic effects. To determine the effects of AZT and AZT-resistant HIV-1 RT on the mutant frequency of HIV-1, the effects of AZT postinfection treatment of target cells infected with AZT-resistant HIV-1 were analyzed. Two AZT-resistant RTs, M41L/T215Y and M41L/D67N/K70R/ T215Y, were initially used in the present study. In parallel experiments, target cells infected with the HIV-1 vector were grown in the presence of 0.4M AZT (Table 1). As indicated in Table 1, the M41L/T215Y RT led to a 3.3-fold increase in the odds of recovering virus mutants, and there was a 5.2-fold increase in the odds with M41L/D67N/K70R/T215Y RT. A drug concentration of 0.4M AZT led to a 6.5-fold increase in the odds of observing virus mutants compared to virus repli-cation with wt HIV-1 RT in the absence of drug. The above increases in the odds of mutant virus recovery are all significant as can be seen from the 95% confidence intervals that do not cover the baseline value of 1. The relative amount of infectious virus produced using M41L/T215Y RT, M41L/D67N/K70R/ T215Y RT, or 0.4M AZT was 60, 40, or 20% of that ob-served for virus replication with wt RT in the absence of drug. If the combination of drug and drug-resistant RT had a multiplicative effect on the odds of virus mutant recovery, an TABLE 1. Multiplicative effect of drugs and drug-resistant RT on HIV-1 mutant frequencies in one round of virus replication
Experimental condition coloniesNo. of No. of mutantcolonies frequencyMutant Relative mutant odds(95% CI)a
AZT and drug-resistant RT
wt 1,368b 8 0.006 1
wt plus 0.4M AZT 1,460 54 0.037 6.5 (3.3–14.9)
M41L/T215Y 942 18 0.019 3.3 (1.5–8.1)
M41L/T215Y plus 0.4M AZT 1,944b 214 0.110 21.0 (11.1–46.6)
M41L/D67N/K70R/T215Y 674 20 0.030 5.2 (2.4–12.6)
M41L/D67N/K70R/T215Y plus 0.4M AZT 1,656b 205 0.124 24.0 (12.6–53.3)
3TC and drug-resistant RT
wt 1,535b 10 0.007 1
wt plus 0.3M 3TC 929 17 0.018 2.8 (1.3–6.5)
M41L/D67N/K70R/T215Y 674 20 0.030 4.7 (2.2–10.4)
M41L/D67N/K70R/T215Y plus 0.3M 3TC 1,656b 136 0.082 13.6 (7.5–27.8)
AZT/3TC and drug-resistant RT
wt 1,939b 11 0.006 1
wt plus 0.4/0.3M AZT/3TC 2,192b 107 0.049 9.0 (5.0–17.8)
M41L/D67N/K70R/T215Y 1,061 20 0.019 3.4 (1.6–7.3)
M41L/D67N/K70R/T215Y plus 0.4/0.3M AZT/3TC 1,476b 191 0.129 26.1 (14.8–51.0)
M41L/D67N/K70R/M184V/T215Y 1,972b 43 0.022 3.9 (2.1–8.0)
M41L/D67N/K70R/M184V/T215Y plus 0.4/0.3M AZT/3TC 1,408b 160 0.114 22.5 (12.7–44.1) HU and drug-resistant RT
wt 1,619b 7 0.004 1
wt plus 2.0 mM HU 1,913b 50 0.026 6.2 (3.0–15.0)
M41L/D67N/K70R/T215Y 1,372 32 0.023 5.5 (2.6–13.6)
M41L/D67N/K70R/T215Y plus 2.0 mM HU 1,551b 134 0.086 21.8 (10.9–51.6)
Thy and drug-resistant RT
wt 2,421b 10 0.004 1
wt plus 50M Thy 1,257 28 0.022 5.5 (2.7–11.9)
M41L/D67N/K70R/T215Y 884 19 0.021 5.3 (2.5–11.9)
M41L/D67N/K70R/T215Y plus 50M Thy 2,204b 143 0.065 16.7 (9.3–34.0)
aColumn shows (odds of mutant colony in group)/(odds of mutant colony in untreated wt group) with 95% confidence intervals. Confidence intervals and significance
statements in the text are based on a logistic model. To test for significant deviations from the model of multiplicative effects of the drug and RT mutants, a statistical test was used for the significance of the interaction term in this model, adjusting for random experiment-to-experiment variation.
bCombined results from two independent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
approximately 21.5-fold increase would be predicted for AZT with M41L/T215Y RT and an approximately 33.8-fold increase in these odds would be predicted for AZT with M41L/D67N/ K70R/T215Y RT. If the drug–drug-resistant RT combination had a synergistic effect on the formation of virus mutants, then the AZT and M41L/T215Y RT or M41L/D67N/K70R/T215Y RT combination would be predicted to result in a fold increase significantly higher than 21.5-fold or 33.8-fold, respectively. An additive effect would have resulted in an approximately 9.4-fold and 11.2-9.4-fold increase in the mutant frequency with the AZT and M41L/T215Y RT or M41L/D67N/K70R/T215Y RT combination, respectively. Finally, if the combined effect of drug and drug-resistant RTs was antagonistic, then the pre-dicted mutant odds would be significantly lower than the indi-vidual increases observed for either AZT, M41L/T215Y RT, or M41L/D67N/K70R/T215Y RT.
Interestingly, virus replication with the M41L/T215Y RT or the M41L/D67N/K70R/T215Y RT in the presence of 0.4M AZT led to 21-fold and 24-fold increases in the odds of virus mutant recovery, respectively, compared to virus replication using wt HIV-1 RT in the absence of AZT (Table 1). The relative amount of infectious virus transfer for either the M41L/T215Y RT or the M41L/D67N/K70R/T215Y RT in the presence of 0.4M AZT was 10% that of virus replication in the absence of drug with wt RT. These levels of infectious virus transfer were slightly lower than the value obtained with wt RT in the presence of 0.4M AZT (which was 20%) but are likely not dramatically lower, as it is expected that the drug-resistant virus would replicate more efficiently in the presence of drug over multiple rounds of replication. Overall, these observations are consistent with a multiplicative effect of drug and drug-resistant RT on the virus mutant frequency. In fact, no signif-icant deviation from the multiplicative model is found (P ⫽
0.59), while an additive model is rejected as an explanation of the data (P⬍0.001).
To determine if other antiretroviral drugs could act together with the AZT resistance mutations in HIV-1 RT to increase the virus mutant frequency, 3TC was tested. In parallel exper-iments, target cells infected with the HIV-1 vector were grown in the presence of 0.3M 3TC (Table 1). In the absence of 3TC, the mutant odds with M41L/D67N/K70R/T215Y RT were about 4.7-fold higher than the mutant odds during rep-lication with wt RT (Table 1). In the presence of 0.3M 3TC, the odds of mutant recovery during one round of replication with the M41L/D67N/K70R/T215Y RT were 13.6-fold higher than those with wt RT in the absence of drug (Table 1). Infectious virus transfer with the M41L/D67N/K70R/T215Y RT in the presence of 3TC was 30% that observed for repli-cation with wt RT in the absence of drug. For an additive or multiplicative effect, the fold increase in mutant odds would have been predicted to be either 7.5- or 13.2-fold, respectively. Therefore, these observations are also consistent (P ⫽ 0.9) with the hypothesis that 3TC acts in a multiplicative manner with AZT-resistant RT during HIV-1 replication and results in corresponding virus mutant frequency increases.
AZT and 3TC treatment together with AZT-resistant or AZT/3TC dually resistant HIV-1 RT can increase the HIV-1
mutant odds in a multiplicative manner.To test whether AZT
and 3TC could act together to influence the HIV-1 mutant frequency with a drug-resistant RT, virus replication with
ei-ther a singly or dually resistant RT in the presence of both drugs was done and the mutant odds were then analyzed. Table 1 shows that AZT/3TC dual treatment during HIV-1 replica-tion with wt RT increased the odds of virus mutants 9-fold compared to that of virus replication in the absence of drug, while AZT/3TC dual treatment with the AZT-resistant M41L/ D67N/K70R/T215Y RT mutant increased the virus mutant odds 26.1-fold over the mutant frequency with wt HIV-1 RT and no drug (Table 1). The 95% confidence interval shows consistency with the hypothesized increase under a multiplica-tive model (predicted odds increase of 9⫻3.4⫽30.6) but not with an additive model (predicted odds increase of 9⫹3.4⫽
12.4). Infectious virus transfer levels for AZT/3TC dual treat-ment alone and for AZT/3TC dual treattreat-ment with the M41L/ D67N/K70R/T215Y RT were both 10% of the level of infec-tious virus transfer observed for virus replication in the absence of drug with wt RT. The virus mutant odds for AZT/ 3TC dual treatment during HIV-1 replication with the AZT/ 3TC dually resistant M41L/D67N/K70R/M184V/T215Y RT mutant led to a 22.5-fold increase in the virus mutant odds compared to replication with wt RT in the absence of drug. In the absence of drug, virus replication with the M41L/D67N/ K70R/M184V/T215Y RT mutant led to an average mutant odds that was about 3.9-fold higher than with virus replication with wt HIV-1 RT and no drug (Table 1). Overall, these results indicate that AZT and 3TC fit the model (P⫽0.37) of acting together to increase HIV-1 mutant recovery along with drug-resistant RT in a multiplicative manner.
HU treatment of cells increases HIV-1 mutant frequency in a dose-dependent manner and acts together with
drug-resis-tant RT to increase mudrug-resis-tant odds.HU, a well-documented drug
used in HIV-1 treatment, is known to alter intracellular dNTP pools by inhibiting ribonucleotide reductase, resulting in a depletion of all dNTPs (3, 8, 9, 11, 21, 31). To determine whether treatment of cells with HU would increase the mutant frequency of HIV-1, the effects of HU postinfection treatment of infected target cells were determined. In parallel experi-ments, target cells infected with the HIV-1 vector were grown in the presence of HU ranging from 0 to 3.0 mM (Fig. 2). As indicated in Fig. 2, HU treatment increased the mutant fre-quency of HIV-1 in a statistically significant dose-dependent manner (P ⬍0.001). The relative amount of infectious virus transfer to target cells compared to controls was reduced to 90% with 1 mM HU, 70% with 1.5 mM HU, 40% with 2.0 mM HU, and 30% with 3.0 mM HU. This indicates that concen-trations of HU that reduced virus transfer by 10% or more meaningfully increased the mutant frequencies to that of the control. The increase in mutant frequency observed with HU posttreatment was interpreted to be due to an increase in the error rate of reverse transcription.
HU treatment was next tested to see if it could enhance the ability of AZT-resistant RT to influence mutant frequencies (Table 1). HIV-1 replication using the M41L/D67N/K70R/ T215Y RT in conjunction with posttreatment of permissive target cells with 2.0 mM HU resulted in a 21.8-fold increase in the odds of virus mutant recovery over that observed in the absence of drug and a 3.5-fold increase in odds over that found for HIV-1 replication with wt RT in the presence of 2.0 mM HU (Table 1). The amount of infectious virus transfer during replication in the presence of HU with the M41L/D67N/K70R/
on November 8, 2019 by guest
http://jvi.asm.org/
T215Y RT was about 10% that of virus replication in the absence of drug with wt RT. In this case the data were consis-tent with the hypothesis of either an additive or a multiplicative effect of AZT-resistant RT and HU.
Thy treatment of cells increases HIV-1 mutant frequency in a dose-dependent manner and acts together with
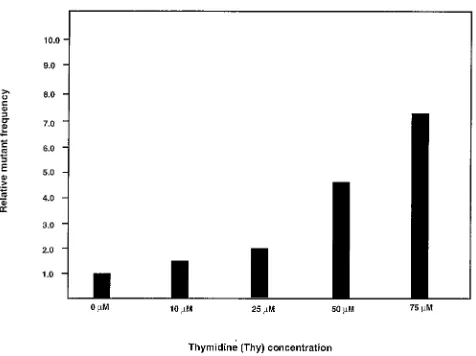
drug-resis-tant RT to further increase virus mudrug-resis-tant odds.Like HU, Thy
has been well documented to alter intracellular dNTP pools and has been shown to increase retrovirus mutation rates (5, 16, 31, 36, 41). To determine whether treatment of cells with Thy would increase the mutant frequency of HIV-1, the effects of Thy treatment of target cells were determined. In parallel experiments, target cells infected with the HIV-1 vector were grown in the presence of Thy, ranging from 0 to 75M (Fig. 3). Posttreatment of cells with 10M Thy resulted in a mutant frequency of 0.008 mutant/cycle, which was 1.6 times higher than the control mutant frequency (0.005 mutant/cycle). Post-treatment of cells with either 25, 50, or 75M Thy resulted in mutant frequencies of 0.010, 0.024, and 0.036 mutant/cycle, respectively. These observations indicate that Thy treatment can increase the mutant frequency of HIV-1 in a statistically significant dose-dependent manner (P⬍ 0.001). The relative amount of infectious virus transfer to target cells compared to controls was reduced to 80% with 25M Thy, 50% with 50M Thy, and 20% with 75M Thy. This indicates that the con-centrations of Thy that reduced virus transfer to 90% or lower meaningfully increased the mutant frequencies to that of the control. The increase in mutant frequency observed with Thy posttreatment, as was observed with HU posttreatment, was interpreted to be due to an increase in the error rate of reverse transcription.
Experiments were then done to determine if Thy treatment could enhance the influence of AZT-resistant RT on the odds of recovering HIV-1 mutants. In particular, virus replication in one round of replication using the M41L/D67N/K70R/T215Y RT mutant was done in conjunction with posttreatment of
permissive target cells with 0.4M Thy (Table 1). The result-ing mutant odds were 16.7-fold higher than the odds of virus mutants with wt RT and no drug and 3-fold higher than the virus mutant odds during HIV-1 replication with the M41L/ D67N/K70R/T215Y RT mutant and no drug. Infectious virus transfer in the presence of Thy with the M41L/D67N/K70R/ T215Y RT was about 10% that observed during replication in the absence of drug with wt RT. The data in this case were consistent with the hypothesis of either an additive or a mul-tiplicative effect of AZT-resistant RT and Thy.
DISCUSSION
Previous work has indicated that antiretroviral drugs or drug resistance mutations in RT can increase the HIV-1 mutation rate. In particular, AZT and AZT-resistant RT alone were found to increase the virus mutation rate by 7.6- and 4.3-fold, respectively (25). The primary objective of this study was to analyze the potential interplay of antiretroviral drugs and drug resistance mutations in RT on their combined effect on HIV-1 mutant frequencies.
[image:5.587.44.285.74.250.2]The effects of various drugs in a single round of replication of AZT-resistant and AZT/3TC dually resistant vector viruses were analyzed. The AZT drug-resistant RTs analyzed, M41L/ T215Y and M41L/D67N/K70R/T215Y, confer high levels of AZT resistance. The AZT/3TC dually resistant RT used, M41L/D67N/K70R/M184V/T215Y, confers AZT and 3TC re-sistance. The drugs studied were AZT, 3TC, HU, and Thy. In general, the increase in the odds of recovering virus mutants observed was approximately multiplicative when drug-resistant virus replication occurred in the presence of drug. The increase in mutant odds was found to range from 13.6-fold (replication of AZT-resistant HIV-1 in the presence of 3TC) to as much as 26.1-fold (replication of AZT/3TC dually resistant HIV-1 in the presence of AZT and 3TC). Thus, each of the drugs tested FIG. 2. HU increases HIV-1 mutant frequencies in a
dose-depen-dent manner. HU was added at the concentrations indicated for 2 h before cocultivation and continuing until 24 h after cocultivation. Analy-sis of the effects of HU at different concentrations was done in parallel and each concentration was tested in at least two independent experi-ments. The average mutant frequency for each HU treatment is shown.
FIG. 3. Thy increases HIV-1 mutant frequencies in a dose-depen-dent manner. Thy was added at the concentrations indicated for 2 h be-fore cocultivation and continuing until 24 h after cocultivation. Analy-sis of the effects of Thy at different concentrations was done in parallel and each concentration was tested in at least two independent experi-ments. The average mutant frequency for each Thy treatment is shown.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.587.303.541.493.671.2]acted together with AZT-resistant RT and increased virus mu-tant frequencies.
Potential mechanisms responsible for increased virus
mu-tant frequencies.The mechanisms for how drugs such as AZT
and 3TC or drug-resistant RTs act to increase virus mutant frequencies are presently being investigated. The observation that the combination of drug-resistant RTs with various drugs led, in general, to a multiplicative increase in the odds of recovering virus mutants suggests that the mechanisms respon-sible for the increase in mutant frequency due to RT with each of the drugs tested could be independent of each other.
Previously, AZT was found to increase the mutation rates of SNV and MLV (15). Hypotheses proposed to explain these increased mutation rates include (i) AZT alters nucleotide pools, (ii) AZT is incorporated into plus-strand DNA and may result in discontinuous DNA synthesis of viral DNAs with proper ends that integrate with subsequent error-prone repair by the host cell, and (iii) AZT may bind noncatalytically to RT and cause a conformational change that influences enzyme fidelity (15). Subsequent work has found that the mechanism for how AZT increases the SNV and MLV mutation rates does not involve alterations of nucleotide pools (16). However, the effect of AZT on nucleotide pools has not been extensively studied in different cell lines or in primary lymphocytes and macrophages, so it is plausible that AZT could influence nu-cleotide pools in particular cell types. One mechanism of AZT resistance has been found to involve the excision of AZT after it has been incorporated (1, 30) and would predict that resistant RT has higher fidelity not lower fidelity. Since AZT-resistant RTs were observed to have lower in vivo fidelity (25), the excision of AZT after incorporation does not increase in vivo fidelity. Also, there have been no reports that indicate that the cell-free fidelity of AZT-resistant RT is enhanced. It is possible that the fidelity for correcting mispairs that do not involve the drug is lower for AZT-resistant RT than for wt RT. Clearly, further analysis of AZT-resistant RT is needed to understand the mechanism(s) responsible for its influence on in vivo fidelity. Like AZT, 3TC is a nucleoside analog and may influence the virus mutation rate by similar mechanisms.
HU and Thy are well documented to alter intracellular dNTP pools. HU alters dNTP pools by inhibiting ribonucle-otide reductase and depleting all dNTPs and may also influ-ence DNA repair by increasing the sensitivity of cells to UV irradiation and to other mutagens. The dNTP pool imbalances induced by Thy treatment likely arise from modulation of the feedback regulation of ribonucleotide reductase. Given that HAART typically combines two or three RT inhibitors for controlling HIV-1 infection, the combined effects of RT on HIV-1 mutant frequencies may be clinically relevant. Further-more, the development of drug resistance in the presence of drug indicates that the combined effect of drugs and drug-resistant RT on HIV-1 mutant frequencies may also be clini-cally relevant.
HIV-1 mutation rate and HIV-1 population dynamics.The
impact of an altered HIV-1 mutation rate is dependent on the population dynamics of the virus population. Deterministic models have been used for predicting the effects of mutation and selection on HIV-1 populations (4, 35), while others have pointed out that stochastic models may be more appropriate (20). A simple “metapopulation” model for HIV-1 replication
shows that the combination of founder effects and subpopula-tion turnover can result in an effective populasubpopula-tion size much lower than the actual population size (7). This lower popula-tion size could contribute to the importance of genetic drift in HIV-1 evolution despite a large number of infected cells. The impact of changes in mutation rate on HIV-1 population dy-namics and evolution needs to be experimentally investigated.
ACKNOWLEDGMENTS
We thank Matt Stachler and Amy Waggoner for technical assis-tance. We also thank an anonymous reviewer for helpful comments and suggestions on the manuscript.
This research was supported by Public Health Service grant GM56615.
REFERENCES
1.Arion, D., N. Kaushik, S. McCormick, G. Borkow, and M. A. Parniak.1998.
Phenotypic mechanism of HIV-1 resistance to 3⬘-azido-3⬘-deoxythymidine
(AZT): increased polymerization processivity and enhanced sensitivity to
pyrophosphate of the mutant viral reverse transcriptase. Biochemistry37:
15908–15917.
2.Bangsberg, D. R., F. M. Hecht, E. D. Charlebois, A. R. Zolopa, M. Holodniy, L. Sheiner, J. D. Bamberger, M. A. Chesney, and A. Moss.2000. Adherence to protease inhibitors, HIV-1 viral load, and development of drug resistance
in an indigent population. AIDS14:357–366.
3.Biron, F., F. Lucht, D. Peyramond, A. Fresard, T. Vallet, F. Nugier, J. Grange, S. Malley, F. Hamedi-Sangsari, and J. Vila.1996. Pilot clinical trial of the combination of hydroxyurea and didanosine in HIV-1 infected
indi-viduals. Antivir. Res.29:111–113.
4.Coffin, J.1995. HIV population dynamicsin vivo: implications for genetic
variation, pathogenesis, and therapy. Science267:483–489.
5.Cohen, A., J. Barankiewicz, H. M. Lederman, and E. W. Gelfand.1983. Purine and pyrimidine metabolism in human T lymphocytes. Regulation of
deoxyribonucleotide metabolism. J. Biol. Chem.258:12334–12340.
6.Finzi, D., M. Hermankova, T. Pierson, L. M. Carruth, C. Buck, R. E. Chaisson, T. C. Quinn, K. Chadwick, J. Margolick, R. Brookmeyer, J. Gal-lant, M. Markowitz, D. D. Ho, D. D. Richman, and R. F. Siliciano.1997. Identification of a reservoir for HIV-1 in patients on highly active
antiret-roviral therapy. Science178:1295–1300.
7.Frost, S. D. W., M.-J. Dumaurier, S. Wain-Hobson, and A. J. Leigh Brown.
2001. Genetic drift and withhost metapopulation dynamics of HIV-1
in-fection. Proc. Natl. Acad. Sci. USA98:6975–6980.
8.Gao, W. Y., A. Cara, R. C. Gallo, and F. Lori.1993. Low levels of deoxynucle-otides in peripheral blood lymphocytes: a strategy to inhibit human
immu-nodeficiency virus type 1 replication. Proc. Natl. Acad. Sci. USA90:8925–
8928.
9.Gao, W. Y., D. G. Johns, and H. Mitsuya.1994. Anti-human
immunodefi-ciency virus type 1 activity of hydroxyurea in combination with 2⬘,3⬘
-dideoxynucleosides. Mol. Pharmacol.46:767–772.
10.Garcia-Lerma, J. G., S. Nidtha, K. Blumoff, H. Weinstock, and W. Heneine.
2001. Increased ability for selection of zidovudine resistance in a distinct class of wild-type HIV-1 from drug-naive persons. Proc. Natl. Acad. Sci.
USA98:13907–13912.
11.Giacca, M., S. Zanussi, M. Comar, C. Simonelli, E. Vaccher, P. de Paoli, and U. Tirelli.1996. Treatment of human immunodeficiency virus infection with
hydroxyurea: virologic and clinical evaluation. J. Infect. Dis.174:204–209.
12.Haubrich, R. H., S. J. Little, J. S. Currier, D. N. Forthal, C. A. Kemper, G. N. Beall, D. Johnson, M. P. Dube, J. Y. Hwang, J. A. McCutchan, et al.1999. The value of patient-reported adherence to antiretroviral therapy in
predict-ing virologic and immunologic response. AIDS18:1099–1107.
13.Hu, W.-S., and H. M. Temin.1990. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of
genetic recombination. Proc. Natl. Acad. Sci. USA87:1556–1560.
14.Ito, W., H. Ishiguro, and Y. Kurosawa.1991. A general method for intro-ducing a series of mutations into cloned DNA using the polymerase chain
reaction Gene102:67–70.
15.Julias, J. G., T. Kim, G. Arnold, and V. K. Pathak.1997. The antiretrovirus
drug 3⬘-azido-3⬘-deoxythymidine increases the retrovirus mutation rate.
J. Virol.71:4254–4263.
16.Julias, J. G., and V. K. Pathak.1998. Deoxyribonucleoside triphosphate pool imbalances in vivo are associated with an increased retroviral mutation rate.
J. Virol.72:7941–7949.
17.Kavlick, M. F., K. Wyvill, R. Yarchoan, and H. Mitsuya.1998. Emergence of multidideoxynucleoside-resistant human immunodeficiency virus type 1 vari-ants, viral sequence variation, and disease progression in patients receiving
antiretroviral chemotherapy. J. Infect. Dis.177:1506–1513.
18.Kuritzkes, D. R.1996. Clinical significance of drug resistance in HIV-1
infection. AIDS10:(Suppl. 5):S27–S33.
on November 8, 2019 by guest
http://jvi.asm.org/
19.Laiken, S. L., C. A. Gross, and P. H. von Hippel.1972. Equilibrium and kinetic studies of Escherichia coli lac repressor-inducer interactions. J. Mol.
Biol.66:143–155.
20.Leigh Brown, A. J.1997. Analysis of HIV-1envgene sequences reveals evidence for a low effective number in the viral population. Proc. Natl. Acad.
Sci. USA94:1862–1865.
21.Lori, F., A. G. Malykh, A. Foli, R. Maserati, A. De Antoni, L. Minoli, D. Padrini, A. Degli Antoni, E. Barchi, H. Jessen, M. A. Wainberg, R. C. Gallo, and J. Lisziewicz.1997. Combination of a drug targeting the cell with a drug targeting the virus controls human immunodeficiency virus type 1 resistance.
AIDS Res. Hum. Retrovir.13:1403–1409.
22.Mansky, L. M.1996. Forward mutation rate of human immunodeficiency
virus type 1 in a T-lymphoid cell line. AIDS Res. Hum. Retrovir.12:307–314.
23.Mansky, L. M.1996. The mutation rate of human immunodeficiency virus
type 1 is influenced by thevprgene. Virology222:391–400.
24.Mansky, L. M.1994. Retroviral-vector-mediated gene transfer, p. 27B:5.1–
27B:5.10.InJ. B. Griffiths, A. Doyle, and D. G. Newell (ed.), Cell and tissue
culture: laboratory procedures, update, 6th ed. John Wiley & Sons, New York, N.Y.
25.Mansky, L. M., and L. C. Bernard.2000. 3⬘-azido-3⬘-deoxythymidine (AZT) and AZT-resistant reverse transcriptase can increase the in vivo mutation
rate of human immunodeficiency virus type 1. J. Virol.74:9532–9539.
26.Mansky, L. M., S. Preveral, E. Le Rouzic, L. C. Bernard, L. Selig, C. Depienne, R. Benarous, and S. Benichou.2001. Interaction of human im-munodeficiency virus type 1 Vpr with the HHR23A DNA repair protein does
not correlate with multiple biological functions of Vpr. Virology282:176–
185.
27.Mansky, L. M., S. Preveral, L. Selig, R. Benarous, and S. Benichou.2000. The interaction of Vpr with uracil DNA glycosylase modulates the human
immunodeficiency virus type 1 in vivo mutation rate. J. Virol.74:7039–7047.
28.Mansky, L. M., and H. M. Temin.1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than predicted from the fidelity of
purified reverse transcriptase. J. Virol.69:5087–5094.
29.Mansky, L. M., and H. M. Temin.1994. Lower mutation rate of bovine
leukemia virus relative to that of spleen necrosis virus. J. Virol.68:494–499.
30.Meyer, P. R., S. E. Matsuura, A. M. Mian, A. G. So, and W. A. Scott.1999. A mechanism of AZT resistance: an increase in nucleotide-dependent
primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell4:35–43.
31.Meyerhans, A., J.-P. Vartanian, C. Hultgren, U. Plikat, A. Karlsson, L. Wang, S. Eriksson, and S. Wain-Hobson.1994. Restriction and enhance-ment of human immunodeficiency virus type 1 replication by modulation of
intracellular deoxynucleoside triphosphate pools. J. Virol.68:535–540.
32.Milich, L., B. Margolin, and R. Swanstrom.1993. V3 loop of the human immunodeficiency virus type 1 Env protein: interpreting sequence variability.
J. Virol.67:5623–5634.
33.Pathak, V. K., and H. M. Temin.1992. 5-azacytidine and RNA secondary
structure increase the retrovirus mutation rate. J. Virol.66:3093–3100.
34.Pillay, D., S. Taylor, and D. D. Richman.2000. Incidence and impact of
resistance against approved antiretroviral drugs. Rev. Med. Virol.10:231–
253.
35.Preston, B. D. 1997. Reverse transcriptase fidelity and HIV-1 variation.
Science275:228–231.
36.Reichard, P.1988. Interactions between deoxyribonucleotide and DNA
syn-thesis. Ann. Rev. Biochem.57:349–374.
37.Richman, D. D.2001. HIV chemotherapy. Nature410:995–1001. 38.Richman, D. D., D. V. Havlir, J. Corbeil, D. Looney, C. C. Ignacio, S. A.
Spector, J. Sullivan, S. Cheeseman, K. Barringer, D. Pauletti, C.-K. Shih, M. Myers, and J. Griffin.1994. Nevirapine resistance mutations of human
im-munodeficiency virus type 1 selected during therapy. J. Virol.68:1660–1666.
39.Schmit, J. C., K. Van Laethem, L. Ruiz, P. Hermans, S. Sprecher, A. Son-nerborg, M. Leal, T. Harrer, B. Clotet, V. Arendt, E. Lissen, M. Witvrouw, J. Desmyter, E. De Clercq, and A. M. Vandamme. 1998. Multiple dideoxynucleoside analogue-resistant (MddNR) HIV-1 strains isolated from
patients from different European countries. AIDS12:2007–2015.
40.Schuurman, R., M. Nijhuis, R. van Leeuwen, P. Schipper, D. de Jong, P. Collis, S. A. Danner, J. Mulder, C. Loveday, C. Christopherson, et al.1995. Rapid changes in human immunodeficiency virus type 1 RNA load and appearance of drug-resistant virus populations in persons treated with
lami-vudine (3TC). J. Infect. Dis.6(Suppl. 3):25–44.
41.Snyder, R. D.1988. Consequences of the depletion of cellular deoxynucleo-side triphosphate pools on the excision-repair process in cultured human
fibroblasts. Mutat. Res.200:193–199.
42.Wong, J. K., M. Hezareh, H. F. Gunthard, D. V. Havlir, C. C. Ignacio, C. A. Spina, and D. D. Richman.1997. Recovery of replication-competent HIV
despite prolonged suppression of plasma viremia. Science278:1291–1295.