Simplex Virus 1 (HSV-1) ICP8 Required for HSV Replication
Kevin F. Bryant,a* Zhipeng Yan,aDavid H. Dreyfus,b* and David M. Knipea
Department of Microbiology and Immunobiology, Harvard Medical School, Boston, Massachusetts, USA,aand Clinical Faculty, Yale School of Medicine, New Haven, Connecticut, USAb
Herpes simplex virus 1 (HSV-1) ICP8 is a single-stranded DNA-binding protein that is necessary for viral DNA replication and
exhibits recombinase activity
in vitro
. Alignment of the HSV-1 ICP8 amino acid sequence with ICP8 homologs from other
her-pesviruses revealed conserved aspartic acid (D) and glutamic acid (E) residues. Amino acid residue D1087 was conserved in every
ICP8 homolog analyzed, indicating that it is likely critical for ICP8 function. We took a genetic approach to investigate the
func-tions of the conserved ICP8 D and E residues in HSV-1 replication. The E1086A D1087A mutant form of ICP8 failed to support
the replication of an ICP8 mutant virus in a complementation assay. E1086A D1087A mutant ICP8 bound DNA, albeit with
re-duced affinity, demonstrating that the protein is not globally misfolded. This mutant form of ICP8 was also recognized by a
con-formation-specific antibody, further indicating that its overall structure was intact. A recombinant virus expressing E1086A
D1087A mutant ICP8 was defective in viral replication, viral DNA synthesis, and late gene expression in Vero cells. A class of
enzymes called DDE recombinases utilize conserved D and E residues to coordinate divalent metal cations in their active sites.
We investigated whether the conserved D and E residues in ICP8 were also required for binding metal cations and found that the
E1086A D1087A mutant form of ICP8 exhibited altered divalent metal binding in an
in vitro
iron-induced cleavage assay. These
results identify a novel divalent metal cation-binding site in ICP8 that is required for ICP8 functions during viral replication.
H
erpes simplex virus 1 (HSV-1) is a double-stranded DNA
virus that replicates its genome in the nuclei of infected cells.
HSV-1 encodes seven gene products that are directly involved in
the replication of viral DNA, all of which are essential for viral
DNA replication (
44
). These proteins are the DNA polymerase
(which consists of U
L30, the catalytic subunit, and U
L42, its
pro-cessivity factor), an origin binding protein (U
L9), a
helicase-pri-mase complex (which consists of the U
L5, U
L52, and U
L8
pro-teins), and the single-stranded DNA binding protein ICP8 (also
known as U
L29), which is the focus of this report.
ICP8 is an early gene product that nonspecifically binds
single-stranded DNA in a cooperative manner (
22
,
38
). In addition to its
essential role in viral DNA replication, ICP8 has also been shown
both to repress transcription from the parental genome (
13
–
15
)
and to stimulate late gene transcription (
12
). ICP8 is known to
bind zinc (
17
), and several biochemical activities of ICP8 require
magnesium cations (
9
,
24
,
34
), suggesting that the protein also
binds magnesium. Zinc binding by ICP8 is thought to be involved
in its DNA binding activity, because altering residues in the ICP8
zinc finger region results in an abolishment of DNA binding (
11
).
The role of magnesium binding by ICP8 is much less well
under-stood, and no mutations have been made that specifically disrupt
ICP8 magnesium binding in order to directly assess its importance
to ICP8 function.
ICP8 has also been reported to mediate several activities
in-volved in DNA recombination
in vitro, including destabilization
of DNA duplexes (
1
), facilitation of the annealing of
single-stranded DNA (
9
), mediation of strand exchange (
2
,
29
,
34
), and
mediation of strand invasion (
28
). ICP8 interacts with the
HSV-encoded alkaline nuclease U
L12, and U
L12 is proposed to play a
role in the initiation and/or the resolution steps of the DNA
re-combination mechanism (
34
,
36
). The interaction of ICP8 with
U
L12 also stimulates the processivity of U
L12-mediated digestion
of DNA (
35
). Other nucleases, such as
Escherichia coli
exonuclease
III and lambda phage Red
␣
exonuclease, can also stimulate the
strand exchange activity of ICP8
in vitro
(
36
). The HSV-1
helicase-primase complex has also been reported to cooperate with ICP8 to
promote strand exchange activity
in vitro
(
29
). Furthermore, ICP8
is required for long-chain DNA synthesis in an
in vitro
recombi-nation-dependent replication assay (
30
,
31
), although it is not
clear that ICP8 recombinase activity is required in this assay. ICP8
is a major component of HSV-1 replication compartments, which
are nuclear domains where viral DNA replication and late gene
expression occur. ICP8 also interacts with several cellular proteins
known to be involved in recombination, including DNA-PKcs
(DNA-dependent protein kinase, catalytic subunit), Rad50, and
Ku86, and recruits these proteins to viral replication
compart-ments (
40
), where they may play important roles in mediating the
recombination of the HSV-1 genome.
In this study, we report the identification of a conserved
aspar-tic acid residue in ICP8 that is required for divalent metal cation
binding and viral DNA replication. ICP8 is thought to bind Mg
2⫹,
and Mg
2⫹is required for ICP8 activity in some
in vitro
recombi-nase assays. Therefore, the conserved D residue in ICP8 may
co-Received13 February 2012 Accepted26 March 2012
Published ahead of print4 April 2012
Address correspondence to David M. Knipe, [email protected].
* Present address: Kevin F. Bryand, Zymo Research Corporation, Irvine, California, USA; David H. Dreyfus, 488 Norton Parkway, New Haven, Connecticut, USA.
We dedicate this article to the memory of Bill Ruyechan, a friend and colleague whose work and advice on ICP8 continues to inspire us.
Supplemental material for this article may be found athttp://jvi.asm.org/.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.00374-12
on November 7, 2019 by guest
http://jvi.asm.org/
ordinate Mg
2⫹, and bound Mg
2⫹at this site may be required for
ICP8 function during HSV-1 infection.
MATERIALS AND METHODS
Cells and viruses.Vero cells were obtained from the American Type Cul-ture Collection (Manassas, VA). The ICP8-complementing cell lines V529 (5) and S2 (11) were generated as described previously. Cells were main-tained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% heat-inactivated fetal bovine serum (FBS) and 5% heat-inacti-vated newborn calf serum (NCS). The growth medium for the V529 and S2 cells was also supplemented with 500g/ml G418.
All experiments were performed with HSV-1 strain KOS as the wild-type (WT) virus (39) or with mutant virus 8lacZ (21), pm1.a (11), or KOS.8DDEm (described below). Viruses were propagated and titrated on Vero or V529 cells by standard procedures.
Plasmids.This section describes the construction of plasmids used in the transient complementation assays. Plasmids used in other assays are described in the relevant sections. Plasmid pFastBac HTa-ICP8 was con-structed by ligating the AvrII/EcoRI fragment containing ICP8 from pSV8.3 (12) into pFastBac HTa (Invitrogen) DNA digested with XbaI and EcoRI (all restriction enzymes were from New England Biolabs [NEB]). The pFastBac HTa-d105 plasmid was constructed in the same manner, except that the AvrII/EcoRI ICP8 fragment originated from pSVd105 (12). The pFastBac HTa-UL29 and pFastBac HTa-UL29.d105 plasmids were constructed by introducing an EcoRI site immediately upstream of theICP8initiation codon in pFastBac ICP8 and pFastBac HTa-d105, respectively, by site-directed mutagenesis, digestion of these plas-mids with EcoRI, and self-ligation to remove the ICP8 5=untranslated region (5=UTR) and vector sequences between the EcoRI site in pFastBac HTa and the ICP8-initiating ATG (newly introduced EcoRI site). pCI⌬.UL29 was constructed by ligating the ICP8-containing DNA frag-ment from pFastBac HTa-UL29 that was digested with HindIII, followed by blunting of the sticky ends with the Klenow fragment ofE. coliDNA polymerase I (NEB) and digestion with EcoRI, into pCI⌬(27) that had been digested with SmaI and EcoRI. pCI⌬.UL29.d105 was constructed in
the same manner, except that the HindIII/Klenow/EcoRI fragment con-taining theICP8gene originated from pFastBac HTa-d105. pCI⌬B was generated by digesting pCI⌬with BamHI, blunting the sticky ends with Klenow fragment, and self-ligating (thus destroying the BamHI restric-tion site). pCI⌬B.UL29 was constructed by ligating the ICP8-containing
DNA fragment from pFastBac HTa-UL29 that was digested with HindIII,
followed by blunting of the sticky ends with Klenow fragment and diges-tion with EcoRI, into pCI⌬B that had been digested with SmaI and EcoRI. Several smaller regions of theICP8gene were subcloned into pBluescript II SK(⫹) (pBS; Stratagene) to be used as the template for introducing D or E codon mutations into theICP8gene by performing PCR-based site-directed mutagenesis. pBS-ICP8-BS contains the BamHI/SalI fragment of theICP8gene cloned into the BamHI and SalI sites in pBS. pBS-ICP8-N contains the NotI fragment of theICP8gene cloned into the NotI site in pBS. The following oligonucleotides, and their complements, were used to generate PCR-based site-specific mutations in theICP8gene (mutated nucleotides are in boldface): for D545A, 5=-ATGAACAGCA TGTACAGCGCCTGCGACGTGCTGGGAAAC-3=; for D547A, 5=-AGC ATGTACAGCGACTGCGCCGTGCTGGGAAACTACGCC-3=; for D625A, 5=-AACGTCAGGCAGGTCGTGGCCCGCGAGGTGGAGCAG CTG-3=; for E627A, 5=-AGGCAGGTCGTGGACCGCGCGGTGGAGCA GCTGATGCGC-3=; for D645A, 5=-GAGGGGAGGAACTTCAAGTTTC GCGCCGGTCTGGGCGAGGCCAACCACGCC-3=; for E735A, 5=-AAA ACGCTGACGGTCGCGCTCTCGGCGGGGGCGGCTATCTGCGCCC CCAGC-3=; for E860A and D861A, 5=-CCGCCCGGCTCCTGTCGCGC GCGGCCATCGAGACCATCGCGTTCAA-3=; for E1086A and D1087A, 5=-GCACCCAGCAGCTGCAGATCGCGGCCTGGCTGGCGCTCCTG GAGGA-3=; for E1086A, 5=-GCACCCAGCAGCTGCAGATCGCGGACT GGCTGGCGCTCCTGGAGGA-3=; for D1087A, 5=-GCACCCAGCAGC TGCAGATCGAGGCCTGGCTGGCGCTCCTGGAGGA-3=; and for
D1087C, 5=-GCACCCAGCTGCAGATCGAGTGCTGGCTGGCGCTCC TGGAGGA-3=.
The D545A, D547A, D625A, and E627A mutations were generated in pBS-ICP8-N, and the AfeI/AgeI DNA fragment from each resulting plas-mid, which contains the mutation, was ligated into AfeI- and AgeI-di-gested pCI⌬.UL29 to generate pCI⌬.UL29.D545A, pCI⌬.UL29.D547A, pCI⌬.UL29.D625A, and pCI⌬.UL29.E627A, respectively. The D645A
mu-tation was generated in pBS-ICP8-BS, and the BamHI/SalI fragment from this plasmid was ligated into BamHI- and SalI-digested pCI⌬B.UL29 to
generate pCI⌬B.UL29.D645A. The E735A, E860A D861A, E1086A
D1087A, E1086A, D1087A, and D1087C mutations were generated in pBS-ICP8-BS, and the AgeI/SalI DNA fragment containing the muta-tions was ligated into AgeI- and SalI-digested pCI⌬.UL29 to generate
pCI⌬.UL29.E735A, pCI⌬.UL29.E860A/D861A, pCI⌬.UL29.E1086A/D1087A,
pCI⌬.UL29.E1086A, pCI⌬.UL29.D1087A, and pCI⌬.UL29.D1087C,
respec-tively. All mutated regions in theICP8gene were confirmed by DNA sequencing.
Transient complementation assay.The transient complementation assay was performed as a variation of our previous studies (33). HeLa cells, which do not complement the replication of the 8lacZ ICP8 mutant virus, were transfected with the indicated plasmids using Effectene transfection reagents (Qiagen) according to the manufacturer’s instructions. At 24 h posttransfection, the transfected cells were infected with 8lacZ virus at a multiplicity of infection (MOI) of 10 PFU per cell. At 24 h postinfection (hpi), samples for determination of viral yield were harvested by scraping the infected-cell monolayer and collecting both the cells and the superna-tant. Samples were frozen at⫺80°C and were thawed, and cell-free super-natant was collected following centrifugation of the samples. The viral yield in each sample was determined by performing plaque assays on V529 cells, which express ICP8 and thus complement the replication of the ICP8 mutant 8lacZ. Complementation was expressed as the percentage of viral yield relative to the viral yield observed in samples transfected with the plasmid expressing wild-type ICP8, which was set at 100% comple-mentation.
Immunoblotting.Vero cells were infected with the indicated virus at an MOI of 10, or HeLa cells were transfected as described above. Cell monolayers were washed with phosphate-buffered saline supplemented with calcium and magnesium (PBS-ABC), and lysates were prepared by scraping the cells in 2⫻sodium dodecyl sulfate-polyacrylamide gel elec-trophoresis (SDS-PAGE) loading buffer and boiling for 5 min. Polypep-tides were resolved by SDS-PAGE and were transferred to a polyvi-nylidene difluoride (PVDF) membrane. Membranes were blocked for 1 h at room temperature with 5% milk in Tris-buffered saline with 0.1% Tween 20 (TBST). Blocked membranes were reacted with primary anti-bodies diluted in 5% milk in TBST. The primary antianti-bodies used were anti-ICP27 (␣-ICP27; Abcam),␣-ICP8 (antibody 3-83 [20]),␣-gC (a gift from Gary Cohen), and anti-glyceraldehyde-3-phosphate dehydrogenase (␣-GAPDH; from either Abcam or Applied Biological Materials Inc.). Horseradish peroxidase-conjugated secondary antibodies were from Santa Cruz Biotechnology.
Generation of recombinant baculoviruses.To generate recombi-nant baculoviruses that express ICP8, we utilized the pFastBac.Dual-based vector (Invitrogen) pFBd.GFP, which expresses green fluores-cent protein (GFP) under the control of the p10 promoter. We obtained pFBd.GFP.S2 (unpublished plasmid), which expresses the reovirus S2 protein, as a gift from Kenneth Murray (Florida Interna-tional University). pFastBac HTa-UL29 and pFastBac
HTa-UL29.E1086A/D1087A were digested with EcoRI and HindIII, and the
DNA fragment containing ICP8 was ligated into pFBd.GFP.S2 that had been digested with EcoRI and HindIII, which removed the S2 coding sequence. The resulting plasmids were named pFBd.GFP.UL29 and
pFBd.GFP.UL29.E1086A/D1087A. ICP8 was epitope tagged at its N
terminus with the 6⫻ His epitope by ligating oligonucleotides encoding six histidine residues into the EcoRI site upstream from the ICP8-initiating ATG codon, generating pFBd.GFP.UL29.His and pFBd.GFP.UL29.E1086A/D1087A.His. The presence of the 6⫻His
on November 7, 2019 by guest
http://jvi.asm.org/
quence in the correct reading frame was confirmed by DNA sequenc-ing. Recombinant baculovirus bacmids were generated by transform-ing chemically competent E. coli DH10Bac cells (Invitrogen) with either pFBd.GFP.UL29.His or pFBd.GFP.UL29.E1086A/D1087A.His
according to the manufacturer’s instructions.
Sf21 insect cells were transfected with recombinant bacmids that ex-press either wild-type ICP8 or the DDE mutant by using the Cellfectin transfection reagent (Invitrogen) according to the manufacturer’s in-structions. The supernatant from transfected cells was collected and was designated the P1 stock. This was used to infect new Sf21 cells and to generate the P2 stock. The P2 stock was used to infect Sf21 cells for the expression and purification of recombinant ICP8 proteins.
Protein purification.Baculovirus-infected Sf21 cells were harvested for protein purification by washing once with PBS, and then cells were collected by scraping from the flask into PBS. Cells were lysed by incubat-ing with a buffer consistincubat-ing of 20 mM Tris (pH 7.5), 300 mM NaCl, 20% glycerol, and 0.5% NP-40 for 30 min on ice. The cell-free supernatant containing the soluble recombinant protein was collected following cen-trifugation of the samples, applied to a column containing Talon affinity resin (Clontech) that was equilibrated with lysis buffer, and allowed to proceed through the column by gravity flow. Bound protein was washed with a buffer consisting of 20 mM Tris HCl (pH 7.5), 300 mM NaCl, 5% glycerol, and 0.1% NP-40 and was then eluted with a buffer consisting of 50 mM Tris HCl (pH 7.5), 300 mM NaCl, 15% glycerol, and 150 mM imidazole. Ten fractions of the eluate were collected. The fractions con-taining the purified protein were identified by resolving a portion by SDS-PAGE and staining the gel with Coomassie stain, and those fractions were combined. The pooled fractions were concentrated, and the buffer was exchanged to ICP8 storage buffer (50 mM Tris HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol [DTT], 20% glycerol) using Vivaspin sample purification columns (Vivascience). Protein concentra-tions were measured using the Bio-Rad protein assay reagent (Bio-Rad) with bovine serum albumin (BSA) dilutions to generate the standard curve.
DNA gel shift assay.A 25-mer oligo(dT) oligonucleotide [oligo(dT)25;
synthesized by Integrated DNA Technologies] was phosphorylated at its 5= end with T4 polynucleotide kinase (NEB) using [␥-32P]ATP (6,000 Ci/mmol,
10 mCi/ml; Perkin-Elmer), and unincorporated radioactive nucleotides were removed using an illustra MicroSpin G-50 column (GE Healthcare). Approx-imately 2 pmol of oligo(dT)25(based on the assumption of 100% recovery
from the MicroSpin column) was incubated on ice with the indicated con-centration of either wild-type ICP8 or the DDE mutant form of ICP8 in ICP8 storage buffer for 30 min. The samples were resolved on a 5% native poly-acrylamide gel, and the gel was dried and exposed to a phosphor storage screen.
Immunofluorescence.Indirect immunofluorescence was performed as described previously (4). Briefly, Vero cells on glass coverslips were either mock infected or infected with the indicated virus at an MOI of 10 PFU/cell for 10 h prior to fixation with 3.65% formaldehyde. The anti-bodies used were 3-83, to detect total ICP8, and the conformation-specific antibody 39S, which detects correctly folded ICP8. The fixed and stained cells were imaged using a Zeiss Axioplan 2 microscope, a Photometrics CoolSNAP HQ2 charge-coupled device (CCD) camera, and Zeiss Axio-Vision 4 image acquisition software.
Construction of mutant viruses.Plasmid p8-8GFP, which encodes ICP8 fused to GFP at its C terminus (41), was linearized by digestion with EcoRI and was cotransfected into S2 cells with 8lacZ infectious DNA to generate the recombinant virus KOS.8GFP. Plaques expressing GFP were identified by fluorescence microscopy, and these recombinant viruses were plaque purified 5 times prior to use in experiments. To generate KOS.8DDEm, KOS.8GFP infectious DNA was cotransfected into V529 cells together with EcoRI-linearized pBS.8flank.8DDEm, which contains the E1086A D1087A mutation inICP8. To generate pBS.8flank.8DDEm, we constructed pBS.8flank by ligating a blunted BglII/NdeI fragment from pSG18 (16), which contains theICP8locus and the surrounding
region, into EcoRV-digested pBS. pBS.8flank.8DDEm was then generated by ligating a Bpu10I fragment containing the E1086A D1087A mutation from pCI⌬.UL29.E1086A/D1087A into Bpu10I-digested pBS-8flank. Plaques that did not express GFP were identified by fluorescence micros-copy and were purified 3 times prior to use in experiments. The presence of the D1086A E1087A mutations in theICP8gene was confirmed by sequencing a PCR product from the appropriate region of ICP8. A recom-binant virus that rescued the ICP8 DDE mutation, KOS.8DDEm-R, was also generated in a similar manner. In this case, infectious KOS.8DDEm DNA was cotransfected into V529 cells together with EcoRI-linearized pBS.8flank. Recombinant viruses were selected by their ability to form plaques on noncomplementing Vero cells and were plaque purified 4 times prior to use in experiments. The presence of wild-type sequence was confirmed by sequencing a PCR product from the appropriate region of ICP8.
Viral yield assay.Vero or V529 cells were infected with the indicated virus at an MOI of 10 in PBS-ABC containing 1% bovine serum and 0.1% glucose for 1 h in a shaking incubator at 37°C. Following the 1-h adsorp-tion step, cells were washed twice with acid wash buffer (40 mM citric acid [pH 3], 10 mM KCl, 135 mM NaCl) and once with DMEM containing 1% FBS, and then DMEM containing 1% FBS was added. Progeny virus was harvested at the indicated times postinfection by scraping the infected-cell monolayer and collecting the cells and supernatant. Samples were frozen at⫺80°C following harvesting. Viral yield was determined by performing plaque assays on V529 cells.
Viral DNA replication assay.Vero cells were infected with the indi-cated virus as described above for the viral yield assay. Following infection for the indicated time, samples were harvested by washing the cell mono-layers with PBS-ABC, and the cells were scraped in PBS-ABC and were then collected by centrifugation. Total DNA (including both cellular and viral DNA) was purified by using the Generation Capture Column kit (Qiagen, formerly Gentra) according to the manufacturer’s instructions. Viral DNA was quantified by performing real-time PCR using primers specific for theICP27promoter (3). Real-time PCR was performed using PowerSYBR green reagents (Applied Biosystems) and an Applied Biosys-tems 7300 sequence detection system according to the manufacturer’s instructions. The viral DNA levels were normalized to the levels of a GAPDHpseudogene in each sample (20).
Iron-induced protein cleavage assay. The iron-induced protein cleavage assay was performed essentially as described previously (19). Briefly, 20 pmol of either wild-type ICP8 or the DDE mutant form of ICP8 was either mock treated or incubated with ammonium iron(II) sulfate hexahydrate at a final concentration of 20M and ascorbic acid at a final concentration of 20 mM, either in the presence or in the absence of 20 pmol of the oligo(dT)25oligonucleotide, in ICP8 storage buffer for 30 min
at room temperature. Hydrogen peroxide was added to the reaction mix-tures treated with ammonium iron(II) sulfate and ascorbic acid, at a final concentration of 0.2%, for 10 s at room temperature. The reactions were stopped by boiling the samples in SDS-PAGE loading buffer, and the polypeptides were resolved by SDS-PAGE, transferred to PVDF mem-branes, and probed for ICP8 using rabbit polyclonal antibody 3-83.
RESULTS
Presence of conserved aspartic acid and glutamic acid residues
in HSV-1 ICP8.
To identify highly conserved regions (and
there-fore potential new functional domains) in HSV-1 ICP8 and its
homologs in other herpesviruses, we performed an alignment of
amino acid sequences from nine ICP8 homologs, with
represen-tatives from alpha-, beta-, and gammaherpesviruses. We
discov-ered numerous aspartic acid (D) and glutamic acid (E) residues
that were conserved in many or all of the ICP8 homologs (some
conserved residues are shown in
Fig. 1
; a complete list is given in
Fig. S1 in the supplemental material), leading us to hypothesize
that these conserved residues are important for ICP8 function.
on November 7, 2019 by guest
http://jvi.asm.org/
Several of these conserved residues were located in or near the
DNA binding groove in the ICP8 crystal structure (
25
), suggesting
that they would be available to carry out enzymatic functions on
bound DNA. Interestingly, members of a family of enzymes called
DDE recombinases, including transposases, RAG-1, and
retrovi-ral integrases, also have conserved D and E residues that
coordi-nate magnesium ions that are important for mediating the
recom-bination reactions, leading us to speculate that ICP8 may share
biochemical and pharmacological properties with these
well-stud-ied proteins. Numerous ICP8 residues were further investigated,
including D545 (amino acid residue positions are based on the
KOS ICP8 sequence [
10
]), D547, D625, E627, D645, E735, E860,
D861, E1086, and D1087.
An ICP8 molecule with an altered conserved D residue has a
decreased ability to support HSV-1 replication.
To determine
whether the conserved D and E residues in ICP8 were required for
ICP8 function during HSV-1 infection, specific D and E codons
were altered to encode alanine in a plasmid encoding HSV-1 ICP8.
The ability of the encoded proteins to complement the replication
of an
ICP8
mutant virus was then determined in a transient
com-plementation assay. All D/E mutant forms of ICP8 were expressed
to similar levels in transfected HeLa cells (
Fig. 2A
). At 24 h after
transfection, the cells were infected with the
ICP8-null mutant
virus 8lacZ at an MOI of 10. Samples were harvested at 24 h
postinfection (hpi), and the viral yield was determined by
per-forming plaque assays on V529 cells, which contain the
ICP8
gene
and can complement the replication of the 8lacZ mutant virus.
The viral yield observed in cells transfected with the plasmid
ex-pressing wild-type ICP8 was designated 100% complementation,
and complementation by the ICP8 mutants was compared to that
value. The d105 mutant form of ICP8, which is not able to
com-plement the replication of an
ICP8
mutant virus (
12
), did not
complement 8lacZ replication to levels above background. The
E860A D861A mutant form of ICP8 complemented 8lacZ
repli-cation to approximately 55% of the level of wild-type ICP8 (
Fig.
FIG 1Alignment of ICP8 homolog sequences. Amino acid sequences for HSV-1 ICP8 and ICP8 homologs from representative viruses in each of the three subfamilies of herpesviruses (alphaherpesviruses, betaherpesviruses, and gammaherpesviruses) were aligned using the T-Coffee alignment algorithm (http://www.tcoffee.org[6]). The ICP8 homologs included in the analysis were from HSV-1 strain KOS (NCBI accession numberP17470), HSV-2 (NP_044499), varicella-zoster virus (AEW89446), Marek’s disease virus (Q9E6P0), Epstein-Barr virus (P03227), human cytomegalovirus (P17147), murine cytomegalovirus (MCMV) (P30672), human herpesvirus 7 (O56282), and Kaposi’s sarcoma-associated herpesvirus (ADQ57880). Sites with similar amino acids in 4 or more ICP8 homologs are in black letters highlighted in light gray; sites with identical amino acids in 4 or more ICP8 homologs are in white letters highlighted in black; and sites with identical amino acids in all 9 ICP8 homologs are in white letters highlighted in dark gray. Two regions are shown, identifying conserved amino acids at positions 545 and 547 (based on their positions in HSV-1 ICP8) and the complete conservation of an aspartic acid residue at position 1087. A full alignment can be found in Fig. S1 in the supplemental material.
FIG 2Transient complementation assay. HeLa cells were either mock trans-fected, transfected with an empty vector (pCI⌬), or transfected with plasmids expressing wild-type ICP8, the ICP8 d105 deletion mutant, or ICP8 with the codons that encode the indicated amino acids mutated to encode alanine. At 24 h posttransfection, the cells were either harvested to prepare samples for immuno-blotting (A) or infected with the ICP8 mutant 8lacZ (B). Viral yield samples were harvested at 24 h postinfection, and viral yield was determined by a plaque assay on ICP8-complementing V529 cells. The reported values are percentages of the com-plementation by cells transfected with the plasmid encoding wild-type ICP8 and are averages for 4 independent experiments. Error bars represent standard devia-tions. Similar results were seen in four additional independent experiments in Vero cells.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.65.516.70.241.2] [image:4.585.300.539.334.610.2]2B
), indicating that residues 860 and 861 are important, but not
essential, for wild-type activity of ICP8. The D645A mutant form
of ICP8 also complemented 8lacZ replication to approximately
50% of the level of wild-type ICP8 (
Fig. 2B
), indicating that
resi-due 645 is also important, but not essential, for wild-type activity
of ICP8. The E1086A D1087A mutant form of ICP8 did not
com-plement the replication of 8lacZ to levels above the background
observed when cells were transfected with the empty-vector
plas-mid (
Fig. 2B
). These results indicated that either residue 1086 or
1087, or both, is absolutely critical for ICP8 to function during
HSV-1 replication in this assay.
To determine whether both residues 1086 and 1087 are
re-quired for ICP8 activity, we changed them each individually to
alanine residues and assayed their abilities to complement 8lacZ
replication. The E1086A mutant form of ICP8 complemented
8lacZ replication to an extent similar to that of wild-type ICP8
(
Fig. 2B
), indicating that this residue is not critical for ICP8
activ-ity. However, the D1087A mutant form of ICP8 did not
comple-ment 8lacZ to levels above background (
Fig. 2B
), indicating that
residue 1087 is absolutely essential for ICP8 to complement the
replication of an
ICP8
mutant virus. Furthermore, the inability of
the E1086A D1087A double point mutant to complement 8lacZ
replication can be attributed entirely to the D1087A mutation.
Cysteine residues are known to be able to coordinate divalent
metal cations in DDE recombinases; therefore, to investigate
whether divalent metal cation binding played a role in the D1087A
phenotype, we tested a mutant form of ICP8 in which residue
D1087 was changed to cysteine. The D1087C mutant form of ICP8
was able to complement 8lacZ to much higher levels than the
D1087A mutant (
Fig. 2B
), suggesting that D1087 coordinates
di-valent metal cations and that this activity is required for ICP8
function. Other amino acid residue mutants (the D545A, D547A,
D625A, E627A, E629A, and E735A mutants) complemented 8lacZ
replication to near-wild-type levels when expressed in HeLa cells
(
Fig. 2B
), indicating that these residues are not absolutely required
for ICP8 function in viral replication.
ICP8 molecules with altered conserved D/E residues are not
globally misfolded.
To rule out that the possibility that the
E1086A D1087A mutation in ICP8 reduced the activity of ICP8 by
simply destroying the overall folding of the protein, we
investi-gated the ability of purified recombinant wild-type and E1086A
D1087A (DDE mutant) ICP8 to bind DNA
in vitro
by performing
electrophoretic mobility shift assays. Both wild-type and DDE
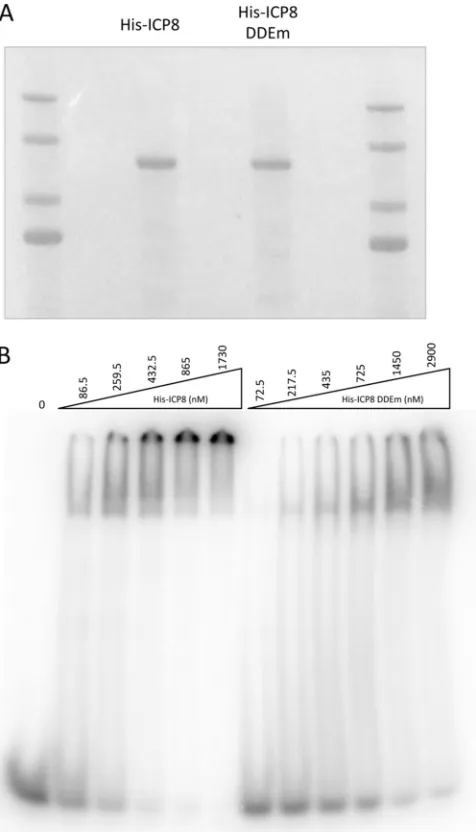
mutant forms of ICP8 were purified to near-homogeneity (
Fig.
3A
). Increasing concentrations of both proteins in the binding
reactions resulted in a slower-migrating form of the labeled
oli-go(dT)
25DNA (
Fig. 3B
), indicating that the DNA was bound by
both forms of purified ICP8 and therefore that the DDE mutant
form of ICP8 is not globally misfolded. However, higher
concen-trations of the DDE mutant form of ICP8 than of wild-type ICP8
were required to bind the oligonucleotide, arguing that the
mu-tant form bound DNA with decreased affinity.
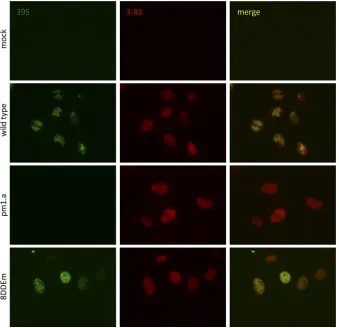
To assess the folding of the mutant form of ICP8 by another
method, we investigated whether mutant ICP8 expressed during
infection with the 8DDEm mutant virus was recognized by the
conformation-specific 39S antibody (
42
). Mock-infected cells
were not stained with either the 39S antibody or the 3-83
␣
-ICP8
antibody, which detects total ICP8, demonstrating the specificity
of these antibodies (
Fig. 4
, top row). Cells infected with wild-type
HSV-1 were stained with both antibodies, and mature replication
compartments were observed (
Fig. 4
, second row). Cells infected
with the
ICP8
mutant virus pm1.a, which expresses an altered
form of ICP8 that is defective for DNA binding and is incorrectly
folded (
11
), were not stained by antibody 39S but were stained by
antibody 3-83 (
Fig. 4
, third row). 8DDEm-infected cells were
stained by both 39S and 3-83 antibodies (
Fig. 4
, bottom row),
further demonstrating that this mutant form of ICP8 maintained
the correct conformation. In addition, we observed that the
mu-tant form of ICP8 expressed during 8DDEm infection did not
form mature replication compartments, indicating that the
al-tered residues were required for this activity.
The conserved D/E residues in ICP8 are required for efficient
HSV-1 replication and viral DNA replication.
We constructed a
FIG 3Effect of ICP8 DDE mutation on DNA binding. (A) His-tagged wild-type and DDE mutant ICP8 proteins were expressed from recombinant bacu-loviruses and were purified from infected Sf21 cells. Purified proteins were resolved by SDS-PAGE, and the gel was stained with Coomassie blue stain. (B) The indicated concentration of each protein was incubated with radiolabeled oligo(dT)25DNA, and protein-DNA complexes were resolved on a 5% native
polyacrylamide gel.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.302.540.67.483.2]mutant virus, 8DDEm, containing the E1086A D1087A mutations
in the
ICP8
gene, to investigate whether this mutant form of ICP8
affected HSV-1 replication when expressed from the viral
ge-nome. We observed that replication of this mutant virus was
de-creased 10- to 100-fold from that with wild-type HSV-1 strain
KOS in noncomplementing Vero cells (
Fig. 5A
). However,
8DDEm replicated better than the
ICP8
mutant virus pm1.a (
Fig.
5A
) (
11
) in Vero cells, suggesting that the replication functions of
the altered ICP8 in the 8DDEm mutant virus were not completely
destroyed. A virus in which the E1086A D1087A mutation was
restored to the wild-type sequence, 8DDEm-R, replicated
simi-larly to the wild-type virus, indicating that the decrease in
replica-tion observed for 8DDEm was likely due to the specific mutareplica-tion.
All viruses replicated to nearly wild type levels in V529 cells, which
express wild-type ICP8 and therefore complement
ICP8
mutant
viruses, suggesting that the replication defects of these viruses in
Vero cells were due to the changes in ICP8.
We also determined the plating efficiencies for the wild-type
virus, the 8DDEm mutant, and the 8DDEm-R rescued virus by
plating serial dilutions of each virus on either noncomplementing
Vero cells or the complementing cell line V529. The wild-type
virus and the rescued virus 8DDEm-R had similar titers on both
cell lines, but the 8DDEm mutant virus had an approximately
70-fold-lower titer on Vero cells than on V529 cells (
Fig. 5B
).
These results are consistent with a replication defect of the
8DDEm mutant virus.
We next investigated the levels of viral DNA replication in Vero
cells infected with either the wild-type virus, the 8DDEm mutant
virus, the 8DDEm-R rescued virus, or the pm1.a
ICP8
mutant
virus, which is defective for DNA binding and viral DNA
replica-tion (
11
). For the wild-type virus, we observed that levels of viral
DNA increased
⬃
1,000-fold during the course of infection (
Fig.
6
). We observed only a very small increase in viral DNA levels
between 4 and 12 h postinfection (
⬃
2-fold), and an
approxi-mately 10-fold increase in viral DNA levels by 24 h postinfection,
with the 8DDEm mutant virus (
Fig. 6
), indicating that DNA
syn-thesis during 8DDEm infection was severely decreased relative to
that of the wild-type virus and strongly suggesting that the E1086
and D1087 residues in ICP8 are essential for efficient HSV-1 DNA
replication. Viral DNA synthesis by the 8DDEm-R rescued virus
was similar to that by the wild type (
Fig. 6
). Viral DNA levels did
not increase at all during the course of infection with pm1.a (
Fig.
6
). Because viral DNA levels in cells infected with 8DDEm were
greater than those in pm1.a-infected cells, it was clear that the
mutant ICP8 expressed by 8DDEm supported low levels of DNA
replication.
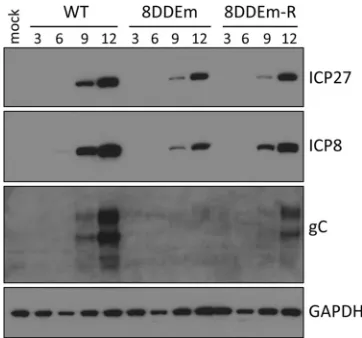
Effect of altered ICP8 D/E residues on viral gene expression.
Because the 8DDEm mutant virus exhibited defects in viral
repli-cation and DNA synthesis, we next investigated whether this
mu-tation also had an effect on viral gene expression. We assayed the
accumulation of the immediate-early
ICP27
gene product, the
early
ICP8
gene product, and the late glycoprotein C (gC) gene
FIG 4Effect of the ICP8 DDE mutation on recognition by the conformation-specific antibody 39S. Vero cells were either mock infected or infected with the indicated virus at an MOI of 10 PFU/cell. At 10 hpi, the cells were fixed and stained with either the␣-ICP8 39S conformation-specific antibody (green; left) or the 3-83 antibody, which detects total ICP8 (red; center).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.124.463.67.395.2]product by performing immunoblot assays with Vero cells that
were infected with either the wild-type virus, the ICP8 mutant
virus 8DDEm, or the rescued virus 8DDEm-R. We observed that
the levels of ICP27 and ICP8 in 8DDEm-infected Vero cells were
slightly lower than those in Vero cells infected with the wild-type
virus but comparable to those in cells infected with the 8DDEm-R
rescued virus (
Fig. 7
), arguing that the E1086/D1087 residues in
ICP8 are not required for the expression of viral immediate-early
or early genes but may be required for maximal levels of
expres-sion. We observed that the late gC protein accumulated to much
lower levels in Vero cells infected with the 8DDEm virus than in
cells infected with the wild-type or 8DDEm-R virus (
Fig. 7
).
Al-though ICP27 expression appeared to be relatively late in this
experiment, this was not observed in other experiments that
con-firmed the reduced expression of gC. In RNA hybridization assays,
we observed patterns of viral transcript accumulation similar to
the patterns of viral protein accumulation seen in immunoblot
assays (results not shown). It is known that gC expression requires
HSV-1 DNA replication (
18
), and the decreased levels of gC are
consistent with the defect in viral DNA replication that we
ob-served.
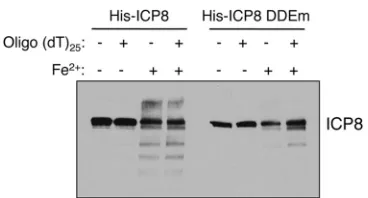
Effect of altered ICP8 D/E residues on divalent cation
bind-ing.
Because D and E residues in DDE recombinases function to
coordinate the binding of magnesium ions (reviewed in reference
7
), we hypothesized that the defect in viral DNA replication with
the 8DDEm mutant virus was due to an altered metal
cation-binding site in ICP8. One approach that has been used to
investi-gate metal-binding sites of DDE recombinases is to measure the
ability of reactive iron ions to cleave proteins when bound (
19
).
Because DDE recombinases coordinate magnesium ions with
their catalytic residues, iron ions can replace magnesium and
pro-mote the cleavage of the bound protein by reactive hydroxyl
rad-icals. This technique was used to successfully map the active-site
residues in RAG1 (
19
), one of the most extensively characterized
DDE recombinases. We observed that purified recombinant ICP8
underwent substantial iron-dependent cleavage in this assay (
Fig.
8
), demonstrating that the wild-type protein bound the iron ion
FIG 5Effects of the ICP8 DDE mutation on viral yield and plating efficiency. (A) Vero and V529 cells were infected at an MOI of 10 PFU/cell with either wild-type HSV-1, the 8DDEm mutant virus, the 8DDEm-R rescued virus, or pm1.a virus. Viral yield samples were harvested at the times indicated, and viral yields were determined by plaque assays on V529 cells. Similar results were seen in two additional independent experiments. (B) Serial dilutions of wild-type HSV-1, the 8DDEm mutant virus, and the 8DDEm-R rescued virus were plated on Vero and V529 cells. The titer of each virus on each cell line is displayed. The values shown are averages for two independent experiments, each performed in duplicate. Error bars represent standard deviations.
FIG 6Effect of the ICP8 DDE mutation on viral DNA synthesis. Vero cells were infected at an MOI of 10 PFU/cell with either wild-type HSV-1, the ICP8 DDEm mutant, the 8DDEm-R rescued virus, or the ICP8 mutant pm1.a, which is defective for DNA binding and replication of viral DNA. Total DNA was harvested at the times indicated, and viral DNA levels in each sample were determined by real-time PCR and were normalized to the levels of cellular DNA. Similar results were seen in two additional independent experiments.
FIG 7Effect of the ICP8 DDE mutation on viral gene expression. Vero cells were infected with either wild-type HSV-1, the ICP8 DDEm mutant, or the 8DDEm-R rescued virus, and lysates were prepared for immunoblotting at the indicated times (hours). Polypeptides were resolved by SDS-PAGE, trans-ferred to a PVDF membrane, and probed for representative immediate-early (ICP27), early (ICP8), and late (gC) gene products.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.300.542.64.210.2] [image:7.585.42.284.68.384.2] [image:7.585.330.511.496.665.2]and therefore suggesting that it is also competent for binding
mag-nesium ions. The amount of iron-dependent cleavage of the
E1086A D1087A mutant form of ICP8 was dramatically reduced
relative to that of the wild-type protein, indicating that this
mu-tant protein was defective for iron ion-induced cleavage and thus
would also be defective for magnesium binding.
DISCUSSION
In this report, we have identified a divalent metal cation-binding
site in HSV-1 ICP8 that is essential for efficient viral replication.
Identification of this site was based on strong conservation with
ICP8 homologs. Plasmids encoding ICP8 with E1086A D1087A
and D1087A mutational alterations failed to complement the
rep-lication of an ICP8-null virus, arguing that the 1087 residue is
essential for ICP8 function and viral replication. A mutant virus,
8DDEm, containing the E1086A D1087A mutations in the
U
L29
gene, which encodes ICP8, exhibits a severe defect in viral
replica-tion and viral DNA synthesis. The mutant virus also shows
re-duced late gene expression. The 8DDEm mutant ICP8 shows
ev-idence of reduced divalent cation binding, consistent with the
notion that this is a DDE recombinase enzymatic domain. ICP8
has been proposed to be a DDE recombinase (
7
,
8
), a member of a
class of enzymes that catalyze recombination using a catalytic triad
of aspartic acid and glutamic acid residues that coordinate
diva-lent metal cations, usually Mg
2⫹. The 8DDEm mutant virus could
provide a new reagent for the functional analysis of ICP8
recom-binase activity during infection.
Nature of the functional defect in 8DDEm ICP8.
ICP8 is a
multifunctional protein that binds to single-stranded DNA,
facil-itates the unwinding of DNA at viral origins of DNA replication,
and interacts with the U
L9 origin-binding protein and the viral
helicase-primase complex to promote viral DNA replication.
Ad-ditionally, ICP8 exhibits recombinase activities
in vitro
(
1
,
2
,
28
–
31
,
34
,
36
). The 8DDEm mutant ICP8 could have defects in one or
more of these activities. The 8DDEm ICP8 is capable of binding to
DNA in a gel shift assay, demonstrating that it is not completely
functionally destroyed. The 8DDEm ICP8 is also recognized by
the conformation-specific 39S monoclonal antibody, evidence
that its conformation and interaction with other HSV DNA
rep-lication proteins are normal (
42
). However, our initial results
in-dicate that the affinity of DNA binding by 8DDEm ICP8 is lower
than that of WT ICP8. Thus, the reduced DNA replication could
be due to reduced DNA binding and promotion of viral DNA
replication. Alternatively, the recombinase function could be
de-fective, and this defect could be responsible for the defect in viral
replication. Further studies of the DNA binding properties and
recombinase activities of this and other ICP8 mutant proteins will
be necessary in order to parse out the precise roles of DNA binding
and recombinase activity in viral replication.
Potential function of ICP8-mediated recombination in
rep-lication.
The recombinase activity of ICP8 could play any of
sev-eral roles in HSV-1 biology. First, recombination could be
in-volved in the circularization of the viral genome upon entry into
the nucleus. However, ICP8 is expressed as a delayed early protein,
and ICP8 has not been demonstrated to be present within the
virion, so it is difficult to postulate a role for ICP8 this early in the
viral replication process. Second, recombination could be
in-volved in the initiation of viral DNA replication or in the
transi-tion from theta-form replicatransi-tion to rolling-circle replicatransi-tion (
37
,
43
). This would be consistent with our observation of a limited
amount of viral DNA replication by the 8DDEm mutant virus.
Third, recombinase activity may also be required to resolve the
branched DNA intermediates that have been observed during
HSV-1 infection, perhaps to generate the linear genomes that are
packaged into virions. In support of this, ICP8 is associated with
progeny viral DNA (
23
). Fourth, the recombinase activity of ICP8
could play a role in the isomerization of the viral genome, which
results in inversion of the genome at the repeated sequences. The
role of genome isomerization is not known; however, viruses in
which the internal repeated sequences are deleted and which are
therefore noninverting retain their ability to replicate (
32
). Fifth,
the ICP8 recombinase activity could lead to conformational
changes in viral progeny DNA that allow efficient late gene
tran-scription.
cis-acting changes in viral genomes following viral DNA
replication are known to activate late gene transcription (
26
).
ICP8 is known to bind to viral progeny DNA (
23
), and the
recom-binase activity of ICP8 could act to resolve concatemers and
in-crease viral late gene transcription. This would be consistent with
the strong defect in gC expression observed with the 8DDEm
mu-tant virus. Finally, general recombination could enhance the
ex-change of genetic information to allow new genotypes that could
be more evolutionarily fit; however, it is not apparent how general
recombination could enhance replication in cultured cells other
than through the roles described above.
Identification of a potential new drug target.
The HIV
inte-grase, a DDE recombinase, has been exploited as a very effective
antiviral target, and multiple small molecules and compounds
that bind the DDE active site and inhibit the activity of this viral
enzyme have been designed. HSV-1 ICP8 may have a DDE active
site similar to the HIV integrase active site, and one of the putative
DDE residues in ICP8 (D1087) was required for efficient HSV-1
DNA replication, suggesting that this site in ICP8 may represent a
novel antiviral target. We are currently working to test this
hy-pothesis and to develop molecules that bind to ICP8 and inhibit its
activity. If these drugs do target a domain including the D1087
resi-due, the conservation of this residue in human alphaherpesviruses
(including HSV-1, HSV-2, and varicella-zoster virus [VZV]), human
betaherpesviruses (including human cytomegalovirus [HCMV] and
human herpesvirus 7 [HHV7]), and human gammaherpesviruses
(including Epstein-Barr virus [EBV] and Kaposi’s
sarcoma-associ-ated herpesvirus [KSHV]) (
Fig. 1
) indicates that this class of drugs
might be applicable to multiple human herpesviruses.
It has been known for several years that ICP8 has recombinase
activity
in vitro, and this study provides evidence that ICP8 shares
FIG 8Effect of the DDE mutation on iron-dependent cleavage of ICP8. Pu-rified His-tagged wild-type ICP8 or ICP8 DDE mutant protein, either in the presence or in the absence of oligo(dT)25DNA, was either mock treated or
treated with the iron/hydroxyl radical reagents. Following the iron-dependent cleavage reaction, samples were resolved by SDS-PAGE, transferred to a PVDF membrane, and probed for ICP8.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.72.256.63.162.2]some similarities with enzymes in the DDE family of
recombi-nases. Additionally, while it is well established that recombination
of the HSV-1 genome occurs at a high frequency, the viral and
cellular factors involved in mediating this recombination, as well
as the role of recombination in viral replication, remain unclear.
This report is the first description of an amino acid residue in ICP8
that may be involved in its recombinase activity, and this study
provides reagents with which to investigate this residue in the
mechanism of ICP8-mediated recombination during HSV-1
rep-lication.
We have identified amino acid residues in ICP8 that are
im-portant for divalent metal cation binding, and we provide
evi-dence that metal ion binding by ICP8 is required for efficient viral
replication. More work is clearly required to define the remaining
residues involved in magnesium binding and to determine
whether these residues constitute a
bona fide
DDE recombinase
active site in ICP8, as well as to define the other proteins that may
be involved in ICP8-dependent recombination. Furthermore, the
precise mechanism of recombination is still unclear, as is the
spe-cific role of recombination during viral DNA replication. The
re-sults in this report provide experimental evidence consistent with
the hypothesis that ICP8 is a DDE recombinase.
ACKNOWLEDGMENTS
This work was funded by NIH grant AI063106 (to D.M.K.). K.F.B. was supported by NIH institutional NRSA training grant AI007245 and NIH individual NRSA fellowship AI081477.
We thank members of the Knipe laboratory for advice and helpful discussions, Kenneth Murray for providing the pFBd.GFP.S2 plasmid, Gary Cohen for providing the␣-gC antibody, and the NERCE Biomol-ecule Production Core Laboratory for providing Sf21 cells and media.
REFERENCES
1.Boehmer PE, Lehman IR.1993. Herpes simplex virus type 1 ICP8: helix-destabilizing properties. J. Virol.67:711–715.
2.Bortner C, Hernandez TR, Lehman IR, Griffith J.1993. Herpes simplex virus 1 single-strand DNA-binding protein (ICP8) will promote homol-ogous pairing and strand transfer. J. Mol. Biol.231:241–250.
3.Bryant K, Colgrove R, Knipe DM.2011. Cellular SNF2H chromatin-remodeling factor promotes herpes simplex virus 1 immediate-early gene expression and replication. mBio 2(1):e00330 –10. doi:10.1128/mBio. 00330 –10.
4.Chang L, et al.2011. Herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late mRNA. Proc. Natl. Acad. Sci. U. S. A.108:E136 –E144.
5.Da Costa XJ, Kramer MF, Zhu J, Brockman MA, Knipe DM. 2000. Construction, phenotypic analysis, and immunogenicity of a UL5/UL29 double deletion mutant of herpes simplex virus 2. J. Virol.74:7963–7971. 6.Di Tommaso P, et al. 2011. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural infor-mation and homology extension. Nucleic Acids Res. 39(Web Server issue):W13–W17.
7.Dreyfus DH.2006. The DDE recombinases: diverse roles in acquired and innate immunity. Ann. Allergy Asthma Immunol.97:567–578, 602. 8.Dreyfus DH.2009. Paleo-immunology: evidence consistent with
inser-tion of a primordial herpes virus-like element in the origins of acquired immunity. PLoS One4:e5778. doi:10.1371/journal.pone.0005778. 9.Dutch RE, Lehman IR. 1993. Renaturation of complementary DNA
strands by herpes simplex virus type 1 ICP8. J. Virol.67:6945– 6949. 10. Gao M, Bouchey J, Curtin K, Knipe DM.1988. Genetic identification of
a portion of the herpes simplex virus ICP8 protein required for DNA-binding. Virology163:319 –329.
11. Gao M, Knipe DM.1989. Genetic evidence for multiple nuclear functions of the herpes simplex virus ICP8 DNA-binding protein. J. Virol.63:5258 – 5267.
12. Gao M, Knipe DM.1991. Potential role for herpes simplex virus ICP8
DNA replication protein in stimulation of late gene expression. J. Virol.
65:2666 –2675.
13. Godowski PJ, Knipe DM.1985. Identification of a herpes simplex virus function that represses late gene expression from parental viral genomes. J. Virol.55:357–365.
14. Godowski PJ, Knipe DM.1983. Mutations in the major DNA-binding protein gene of herpes simplex virus type 1 result in increased levels of viral gene expression. J. Virol.47:478 – 486.
15. Godowski PJ, Knipe DM.1986. Transcriptional control of herpesvirus gene expression: gene functions required for positive and negative regu-lation. Proc. Natl. Acad. Sci. U. S. A.83:256 –260.
16. Goldin AL, Sandri-Goldin RM, Levine M, Glorioso JC.1981. Cloning of herpes simplex virus type 1 sequences representing the whole genome. J. Virol.38:50 –58.
17. Gupte SS, Olson JW, Ruyechan WT.1991. The major herpes simplex virus type-1 DNA-binding protein is a zinc metalloprotein. J. Biol. Chem.
266:11413–11416.
18. Jones PC, Roizman B.1979. Regulation of herpesvirus macromolecular synthesis. VIII. The transcription program consists of three phases during which both extent of transcription and accumulation of RNA in the cyto-plasm are regulated. J. Virol.31:299 –314.
19. Kim DR, Dai Y, Mundy CL, Yang W, Oettinger MA.1999. Mutations of acidic residues in RAG1 define the active site of the V(D)J recombinase. Genes Dev.13:3070 –3080.
20. Knipe DM, Senechek D, Rice SA, Smith JL.1987. Stages in the nuclear association of the herpes simplex virus transcriptional activator protein ICP4. J. Virol.61:276 –284.
21. LaVail JH, et al.2005. Genetic and molecular in vivo analysis of herpes simplex virus assembly in murine visual system neurons. J. Virol.79: 11142–11150.
22. Lee CK, Knipe DM.1985. An immunoassay for the study of DNA-binding activities of herpes simplex virus protein ICP8. J. Virol.54:731– 738.
23. Lee CK, Knipe DM.1983. Thermolabile in vivo DNA-binding activity associated with a protein encoded by mutants of herpes simplex virus type 1. J. Virol.46:909 –919.
24. Makhov AM, Griffith JD.2006. Visualization of the annealing of com-plementary single-stranded DNA catalyzed by the herpes simplex virus type 1 ICP8 SSB/recombinase. J. Mol. Biol.355:911–922.
25. Mapelli M, Panjikar S, Tucker PA.2005. The crystal structure of the herpes simplex virus 1 ssDNA-binding protein suggests the structural ba-sis for flexible, cooperative single-stranded DNA binding. J. Biol. Chem.
280:2990 –2997.
26. Mavromara-Nazos P, Roizman B.1989. Delineation of regulatory do-mains of early (beta) and late (gamma 2) genes by construction of chime-ric genes expressed in herpes simplex virus 1 genomes. Proc. Natl. Acad. Sci. U. S. A.86:4071– 4075.
27. Murphy CG, et al.2000. Vaccine protection against simian immunode-ficiency virus by recombinant strains of herpes simplex virus. J. Virol.
74:7745–7754.
28. Nimonkar AV, Boehmer PE. 2003. The herpes simplex virus type-1 single-strand DNA-binding protein (ICP8) promotes strand invasion. J. Biol. Chem.278:9678 –9682.
29. Nimonkar AV, Boehmer PE.2002. In vitro strand exchange promoted by the herpes simplex virus type-1 single strand DNA-binding protein (ICP8) and DNA helicase-primase. J. Biol. Chem.277:15182–15189. 30. Nimonkar AV, Boehmer PE.2003. Reconstitution of
recombination-dependent DNA synthesis in herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A.100:10201–10206.
31. Nimonkar AV, Boehmer PE.2004. Role of protein-protein interactions during herpes simplex virus type 1 recombination-dependent replication. J. Biol. Chem.279:21957–21965.
32. Poffenberger KL, Tabares E, Roizman B.1983. Characterization of a viable, noninverting herpes simplex virus 1 genome derived by insertion and deletion of sequences at the junction of components L and S. Proc. Natl. Acad. Sci. U. S. A.80:2690 –2694.
33. Quinlan MP, Knipe DM.1985. Stimulation of expression of a herpes simplex virus DNA-binding protein by two viral functions. Mol. Cell. Biol.
5:957–963.
34. Reuven NB, Staire AE, Myers RS, Weller SK.2003. The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J. Virol.77:7425–7433.
35. Reuven NB, Weller SK.2005. Herpes simplex virus type 1
on November 7, 2019 by guest
http://jvi.asm.org/
strand DNA binding protein ICP8 enhances the nuclease activity of the UL12 alkaline nuclease by increasing its processivity. J. Virol.79:9356 – 9358.
36. Reuven NB, Willcox S, Griffith JD, Weller SK.2004. Catalysis of strand exchange by the HSV-1 UL12 and ICP8 proteins: potent ICP8 recombi-nase activity is revealed upon resection of dsDNA substrate by nuclease. J. Mol. Biol.342:57–71.
37. Roizman B, Jacob RJ, Knipe DM, Morse LS, Ruyechan WT.1979. On the structure, functional equivalence, and replication of the four arrange-ments of herpes simplex virus DNA. Cold Spring Harb. Symp. Quant Biol.
43(Pt. 2):809 – 826.
38. Ruyechan WT.1983. The major herpes simplex virus DNA-binding pro-tein holds single-stranded DNA in an extended configuration. J. Virol.
46:661– 666.
39. Schaffer P, Vonka V, Lewis R, Benyesh-Melnick M.1970. Temperature-sensitive mutants of herpes simplex virus. Virology42:1144 –1146.
40. Taylor TJ, Knipe DM.2004. Proteomics of herpes simplex virus replica-tion compartments: associareplica-tion of cellular DNA replicareplica-tion, repair, re-combination, and chromatin remodeling proteins with ICP8. J. Virol.
78:5856 –5866.
41. Taylor TJ, McNamee EE, Day C, Knipe DM.2003. Herpes simplex virus replication compartments can form by coalescence of smaller compart-ments. Virology309:232–247.
42. Uprichard SL, Knipe DM.2003. Conformational changes in the herpes simplex virus ICP8 DNA-binding protein coincident with assembly in viral replication structures. J. Virol.77:7467–7476.
43. Ward SA, Weller SK.2011. HSV-1 DNA replication, p 89 –112.InWeller SK (ed), Alphaherpesviruses: molecular virology. Caister Academic Press, Norfolk, United Kingdom.
44. Wu CA, Nelson NJ, McGeoch DJ, Challberg MD.1988. Identification of herpes simplex virus type 1 genes required for origin-dependent DNA synthesis. J. Virol.62:435– 443.
on November 7, 2019 by guest
http://jvi.asm.org/